Ron Rosenfeld Pediatric Research Lab
Insulin-like growth Factor Deficiency (IGFD) in children with severe growth failure:
defects in the growth hormone (GH)-IGF-I axis.
Growth is one of the most fundamental, complex and poorly understood of biological processes. Over the last decade, however, the combination of targeted gene deletion studies in rodents and human mutational analysis has clearly demonstrated the primary role of the IGFs (insulin-like growth factor) in both intrauterine and postnatal growth. Our laboratory has a long-standing interest in growth failure resulting from defects in IGF-I production and action. As part of this focus, we have established an IGF Deficiency Research Center. Elucidating the mechanisms by which IGF-I production and actions are modulated should provide valuable insights to the fundamental mechanisms involved in human growth and, ultimately, will have important implications in the management of growth disorders.
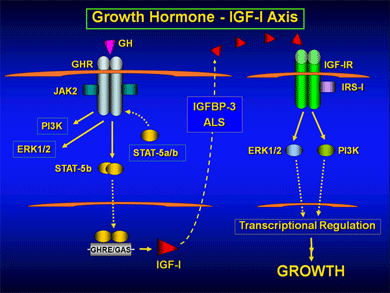
Role of GH-IGF-I axis in human growth
Growth in humans is characterized by a number of unique features, including dramatic fetal growth, deceleration of growth immediately after birth, a prolonged growth phase during childhood, and a pronounced adolescent growth spurt. Studies to date indicate that the insulin-like growth factor (IGF) system is critical for all phases of mammalian growth. During postnatal life, IGF-I production is most dependent on growth hormone (GH), although other growth factors and cytokines can also regulate IGF-I synthesis. Disturbances in IGF-I production or action, therefore, lead to poor linear growth, as well as other developmental aberrations.
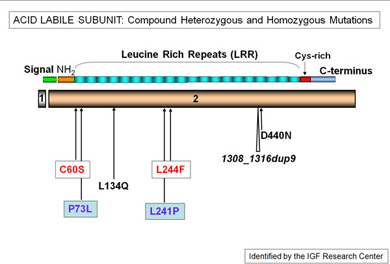
IGF-I circulates in serum as part of a ternary complex with IGF binding protein (IGFBP)-3 and acid labile subunit (ALS), both of which are also subject to regulation by GH. Abnormally low levels of circulating IGF-I may result from GH deficiency (secondary IGFD) or can be indicative of primary IGFD, with severe growth retardation in the presence of normal GH levels (GH insensitivity, GHI). GH action is initiated upon binding of GH to the GH receptor (GHR), which activates the JAK-STAT (Janus kinase-signal transducer and activators of transcription), PI3K (phosphatidylinsolitol-3 kinase) and MAPK (mitogen-activated protein kinase) signaling pathways, and cumulates in the regulation of multiple growth promoting genes, including IGF-I, IGFBP-3, and ALS.
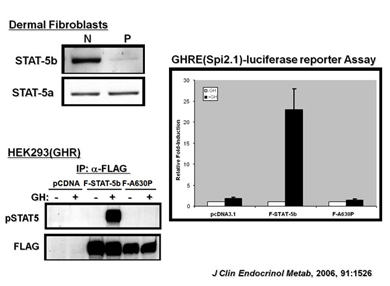
We are interested in abnormalities along the human GH-IGF-I axis that results in a clinical phenotype of severe growth failure, with the focus on understanding the molecular mechanism(s) leading to primary IGF deficiency (IGFD) and the syndrome of GH insensitivity (GHI). To this end, we identified the first case of GHI/IGFD due to a mutation in the STAT5b gene (New England Journal of Medicine, 2003, 349:1139-1147), and have since identified 3 additional cases of IGFD/GHI due to autosomal, recessive, STAT5b mutations.
Recent mutations identified in patients with abnormal growth profiles include defects in the GHR, the ALS, and the IGF-I receptor (IGFIR) genes. Through reconstitution studies complemented by investigations utilizing primary dermal fibroblasts established from the subjects, valuable insights are gained regarding how these aberrant proteins result in poor human growth. The combination of clinical and basic research should advance our understanding of normal and abnormal growth in humans.