Pediatric Cystic Fibrosis

Cystic fibrosis is a disease that mainly affects the lungs and digestive system. Children with the disease are prone to lung infections and other complications. At OHSU Doernbecher Children’s Hospital, our doctors and other specialists are dedicated to helping your child or teen grow normally. We offer you:
- Oregon’s only nationally accredited Cystic Fibrosis Care Center staffed by pediatric pulmonologists.
- Trikafta, a medication approved by the U.S. Food and Drug Administration in 2019 that's showing dramatic results.
- Coordinated care from diagnosis through treatment and monitoring.
- A family council to improve your child’s care experience.
- A team of child specialists, working together to care for your child.
Complete care
The specialists at Doernbecher’s Cystic Fibrosis Care Center are experts at thinning mucus, supporting nutrition and treating infections. We will be your partner in care decisions for your child. Our services and expertise include:
World-class pediatric cystic fibrosis specialists: Our pulmonologists are experts in treating and managing complex cystic fibrosis conditions in children and teens.
A full team of specialists that may include:
- A pulmonologist
- A respiratory therapist
- Nurses
- A dietitian
- An otolaryngologist (ear, nose and throat surgeon)
- Gastroenterologist (digestive system doctor)
- Speech-language therapist
- Social worker
- Pharmacist
A dedicated lab for children: Our respiratory therapists have the latest pulmonary function testing technology to measure and monitor your child’s lungs. Your child’s care team has immediate access to the test results in your child’s electronic medical records.
Same-day testing: Your child’s tests and appointments with a variety of specialists are scheduled on the same day.
Leading-edge sleep medicine: Doernbecher does sleep tests in a homelike setting where you can stay with your child.
Cystic Fibrosis Family Council: This advisory group works to improve the care experience for people with cystic fibrosis. The group is made up of parents and families that meet monthly with our medical team to discuss care, quality improvement and Family Education Day.
Cystic Fibrosis Family Education Day: This event provides updates on advances in research. Families can also meet with one another and learn about care approaches for respiratory therapy and nutrition. See the Learn more section below for links to presentations.
Understanding cystic fibrosis
What is cystic fibrosis?
Cystic fibrosis is an inherited disorder that causes repeated lung infections, digestive problems and other complications. It is progressive, meaning it worsens over time.
Children with cystic fibrosis have an abnormal gene that results in a defective protein. The protein (cystic fibrosis transmembrane conductance regulator, or CFTR) disrupts the balance of chloride, water and salt (sodium) needed to hydrate cells. This results in a buildup of thick, sticky mucus in your child’s bronchial tubes (airways), pancreas, intestines and liver.
Cystic Fibrosis
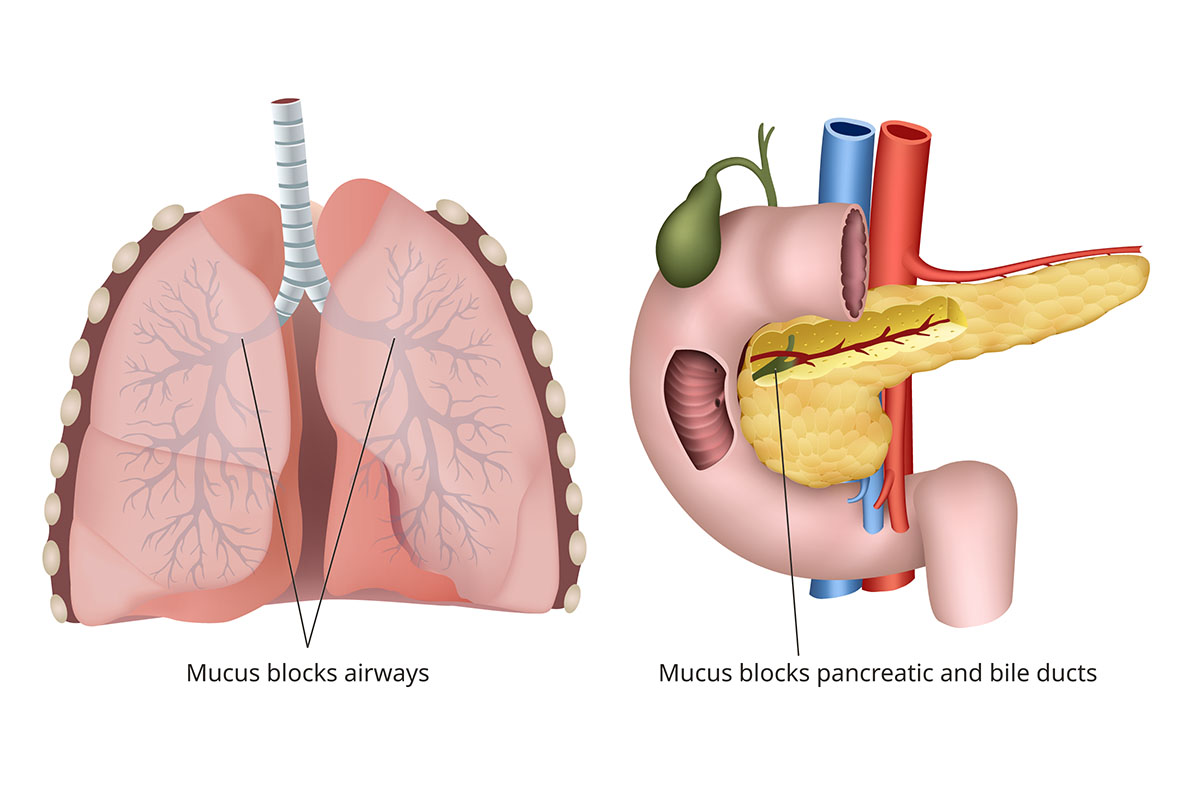
Breathing: The mucus clogs airways, making it more difficult for your child to breathe. The mucus also traps bacteria in the lungs and airways, resulting in inflammation and infections. Over time, this can injure the lungs.
Digestion: In the pancreas, the mucus blocks digestive enzymes from entering the intestines to help break down food. This reduces the nutrients and calories your child needs to gain weight and grow normally. Damage to the pancreas can lead to cystic fibrosis-related diabetes.
Fertility: Most males with cystic fibrosis have fertility issues because the vas deferens, the tube that carries sperm to the penis, does not develop properly.
Who gets cystic fibrosis?
Children inherit the disease from their parents. The mother and father each have to carry one abnormal cystic fibrosis gene. It takes two copies for a child to have cystic fibrosis. When two carriers of the abnormal gene have a child, there’s a one in four chance the child will have cystic fibrosis.
More than 30,000 people in the U.S. are living with cystic fibrosis, according to the Cystic Fibrosis Foundation, and about 1,000 cases are diagnosed each year.
While there is no cure for cystic fibrosis, treatment advances have increased life expectancy. Most patients now live into their 40s. With potential gene therapies being developed to target the defective gene, a baby born today could live longer.
Symptoms
Symptoms include:
- Salty skin and sweat
- Chronic cough with discolored mucus
- Frequent lung infections
- Wheezing or asthma-like symptoms
- Nasal polyps — growths in the nasal passages and sinuses
- Poor weight gain and growth
- Unpleasant-smelling stools
- Intestinal blockage
Complications
Over time, cystic fibrosis may cause:
- Lung damage
- Cystic fibrosis-related diabetes
- Liver disease
- Osteoporosis (bone disease)
- Anxiety and depression
Diagnosing cystic fibrosis
Newborn screening: A blood test on a sample from your newborn is 93% accurate. Most infants with cystic fibrosis are diagnosed this way.
Sweat chloride test: This painless test measures the amount of salt in sweat to confirm a positive newborn screening. Newborns and children with cystic fibrosis have a higher level of salt (sodium and chloride) in their sweat.
Prenatal screening: Your obstetrician can do two genetic tests while you’re pregnant to find out if your baby has cystic fibrosis:
- Chorionic villus sampling: A tiny sample of placenta is removed for testing.
- Amniocentesis: A sample of amniotic fluid, the fluid around your baby, is removed with a hollow needle for testing.
Carrier screening: A blood test can determine if a person carries the cystic fibrosis gene.
Cystic fibrosis treatments
Treating and managing a complex disease such as cystic fibrosis requires therapies for many parts of your child’s body. At Doernbecher, your child’s providers will work together to coordinate care.
-
Manual chest percussion for infants: In this therapy, you tap on your baby’s ribs to ease breathing.
Positive expiratory pressure: Your child breathes through a device that provides resistance when exhaling. This helps air get behind mucus and push it out of the lungs and airways.
Huff breathing: In this technique, your child takes a tiny breath in and slowly exhales. It helps move mucus from deep in the lungs.
Vest therapy (high-frequency chest wall oscillation): A vest connected to an air pulse generator rapidly inflates and deflates to loosen mucus so it can be coughed up. Your child typically uses the vest twice a day for 20 to 30 minutes at a time.
-
Trikafta: Doernbecher offers Trikafta, a medication approved in 2019 for children ages 12 and older who have the most common form of cystic fibrosis.
This three-medication combination, given in daily pills, treats the gene mutation that affects about 90% of cystic fibrosis patients. It’s considered the biggest breakthrough for cystic fibrosis since the gene that causes it was found in 1989.
Other daily medications and supplements may be prescribed. They include:
- Inhaled short-acting bronchodilators (such as albuterol) to keep airways open.
- Mucus-thinning drugs to help with coughing up mucus.
- Antibiotics to treat and prevent lung infections.
- Anti-inflammatory medications to reduce swelling in airways.
- Pancreatic enzyme tablets to improve digestion and help the body absorb fat and nutrients.
- Vitamin supplements including vitamin D for strong bones.
- Cystic fibrosis transmembrane conductance gene therapy (Orkambi and Kalydeco) in pill form to target specific defects.
-
A cystic fibrosis dietitian works with you to create meal plans to make sure your child gets enough calories and nutrients. The dietitian will help track your child’s growth and weight gain.
-
A team of social workers screens your child for anxiety and depression. They also work with families and patients to provide emotional support. Teenagers and young adults are at the highest risk of being sad and worried.
Living with cystic fibrosis
Your child will have regular visits with a care team:
- Ages 1 month to 6 months: You and your newborn meet with your pulmonology doctor every month.
- Ages 6 months to 1 year: You have appointments every two months.
- Age 1 and beyond: Appointments are every three months to monitor and adjust your child’s treatment.
Visits include:
- Tracking your child’s weight and height.
- Breathing tests to measure how the lungs are working.
- Annual blood tests.
- Chest X-ray or CT scan if your child has respiratory infection symptoms.
- Prescribing any needed medications or treatments.
Oral glucose tolerance test: Starting at age 10, your child will have this annual test for cystic fibrosis-related diabetes.
Daily breathing treatments: Each day, your child will do some form of airway clearing to help rid thick mucus from the lungs.
Inhaled medication: Your child will use a device called a nebulizer to inhale a mist of antibiotics and short-acting bronchodilators. This helps to keep airways clear and prevent lung infections.
Oral medication/supplements: Your child will take daily pills that may include:
- Enzyme supplements
- Multivitamins
- Antibiotics
- Anti-inflammatories
- Mucolytics (mucus-thinning medications)
- Cystic fibrosis transmembrane conductance regulator gene therapy
Nutrition: Your child will need a diet high in calories and protein to grow and maintain weight. You can add extra calories to favorite foods, such as by adding avocado, bacon and cheese to a sandwich. You can also give your child high-calorie drinks such as whole milk.
Learn more
Cystic Fibrosis Family Education Day
-
- Welcome | Michael Powers, M.D.; Kelvin MacDonald, M.D.; and Corinne Muirhead, Pharm.D.
- Review of CFLN | Michael Powers, M.D.; Family Advisory Council video; Ben McCullar, RN; Emily Somervell, LCSW; Patricia Rose, RD
- CF Interactive Session| Michael Powers, M.D.
- Respiratory Therapy Education for Young Children | Kim Keeling, RT
- Respiratory Therapy Education for Teens | Kim Keeling, RT
- Mental Health Screening/Center Data | Alexandra Tharp, MSW, CSWA; Michael Powers, M.D.
- Research Updates and “A Day in the Life” of a Research Participant | Brendan Klein, M.P.H., CCRP
- Next Gen Modulators/Next Ten Years | Michael Powers, M.D.
-
- Cystic Fibrosis Learning Network | Michael Powers, M.D., and Lindsay Deveaux
- Partnerships for Sustaining Daily Care | Cindy George, M.S.N., FNP, and Katherine Raymond, M.S.W.
- Equipment Cleaning/PEP | Kim Keeling, RT
- CF Cultures: Simplified! | Kelvin MacDonald, M.D.
- Research Pipeline/OHSU Research/Clinical Trial Finder | Michael Powers, M.D., and Brendan Klein, M.P.H., CCRP
- CF 103 | Michael Powers, M.D., and Megan Johnson, RD
- Cystic Fibrosis Center Data | Michael Powers, M.D.
More information
- OHSU clinical trials
- Cystic Fibrosis, National Heart, Lung, and Blood Institute
- Cystic Fibrosis Foundation
- OHSU Cystic Fibrosis Research Team
- Cystic Fibrosis News Today
- Cystic Fibrosis, Genetics Home Reference
Location
Parking is free for patients and their visitors.
Doernbecher Children’s Hospital
700 S.W. Campus Drive
Portland, OR 97239
Map and directions
Refer a patient
- Refer your patient to OHSU Doernbecher.
- Call 503-346-0644 to seek provider-to-provider advice.