Skull Anomaly (Craniosynostosis)
The specialists at OHSU Doernbecher Children’s Hospital are national leaders in diagnosing and treating craniosynostosis. This condition causes an infant’s skull to grow in an unusual shape.
- We do 60 to 80 surgeries a year, more than any other medical center in Oregon. Our expertise attracts patients from across the West.
- Our team approach means you can see all of your child’s specialists in one day.
- Our team meets often to talk about your child’s care, making sure your child receives the best possible treatment.
- We make sure your child receives steady care over the years it takes to treat their condition. This includes regular follow-up visits for monitoring and treatment.
Understanding craniosynostosis
In craniosynostosis, bones in a baby’s skull fuse too early. The head may not grow as it should.
It’s important to know that craniosynostosis is treatable. Outcomes are often excellent, especially when treatment starts early.
Not all changes in head shape are craniosynostosis. A common condition called plagiocephaly, with flattening, can result from lying in one position. It improves over time or with simple treatment. Your OHSU care team can give you a precise diagnosis.
Normal development
To understand craniosynostosis (from words meaning skull, bone and together), it helps to understand normal development.
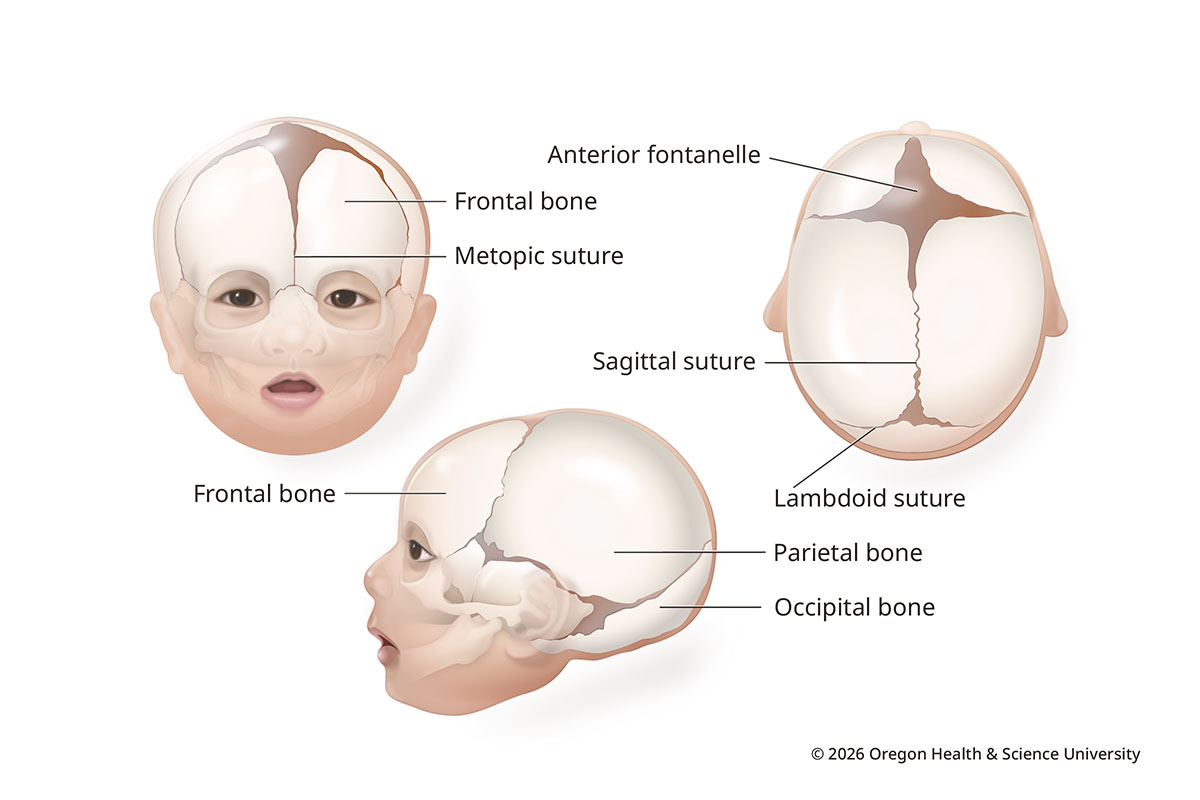
A newborn’s skull isn’t solid like an adult’s. It has bone plates connected with strong, flexible tissue called sutures. It also has soft spots called fontanelles between sections of bone.
This anatomy lets the head expand as the brain grows. The soft spots and sutures then close over time. Soft spots usually close by 18 months, sutures by the late teens or adulthood.
Normal Infant Skull
What is craniosynostosis?
In craniosynostosis, one or more of the sutures closes early. This limits how the head can grow, affecting shape or symmetry. In some cases, brain development may be affected.
Craniosynostosis affects about 1 in 2,000 to 2,500 newborns in the U.S. each year. It may not be noticeable until the baby grows.
What causes craniosynostosis?
Doctors usually don’t know. Genetic or environmental factors can play a role. Having a sibling or parent with craniosynostosis increases risk, for example. Most cases happen by chance, not because of anything a parent did or didn’t do.
Types of craniosynostosis
Sagittal synostosis (scaphocephaly)
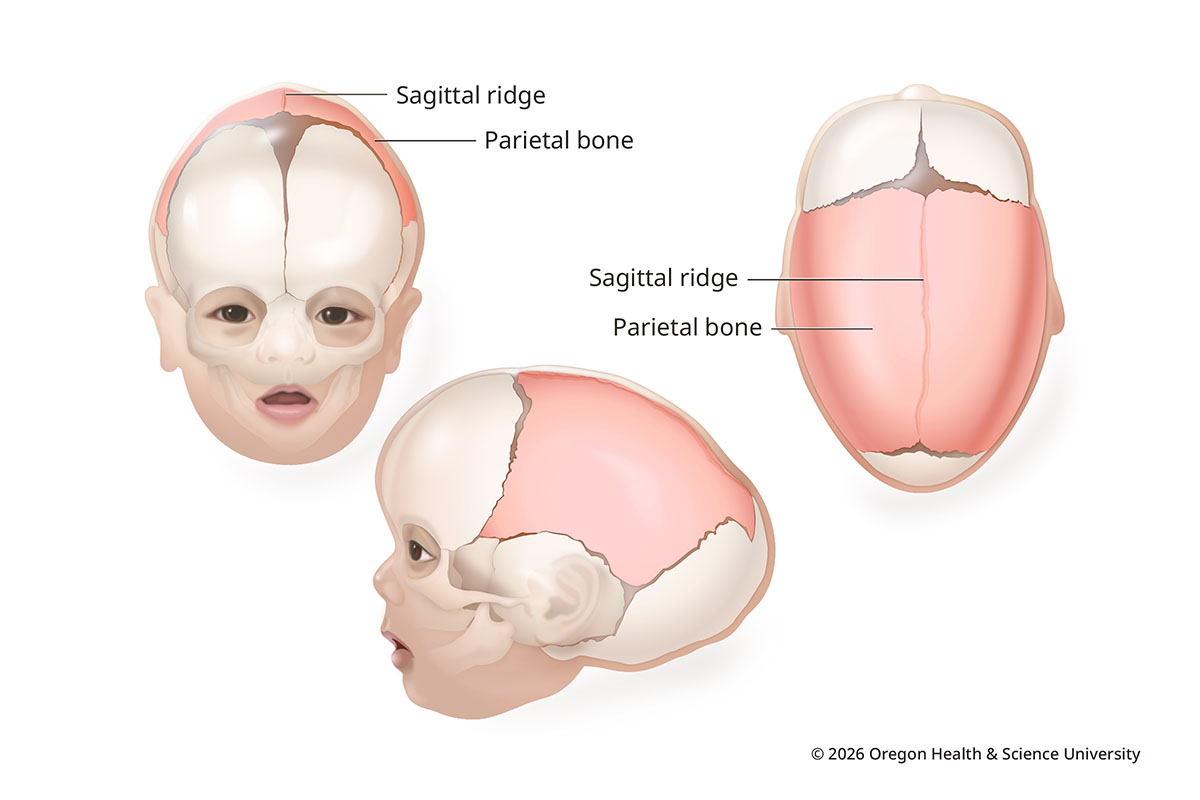
Sagittal synostosis is the most common type of craniosynostosis. It makes up about 40-60% of cases. It happens when the seam at the middle of the skull closes early.
The head grows long and narrow. The forehead and the back of the head look more prominent. Bone forms a ridge over the seam.
Sagittal Synostosis
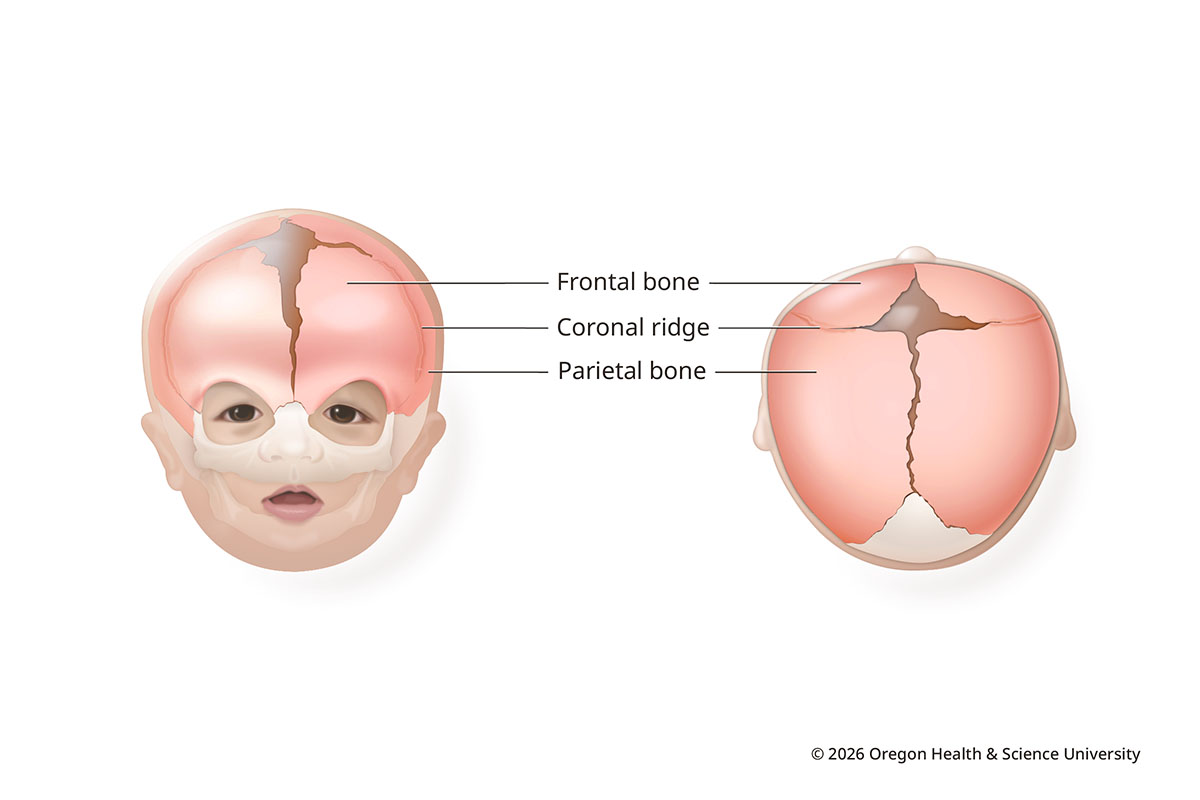
Coronal synostosis (anterior plagiocephaly)
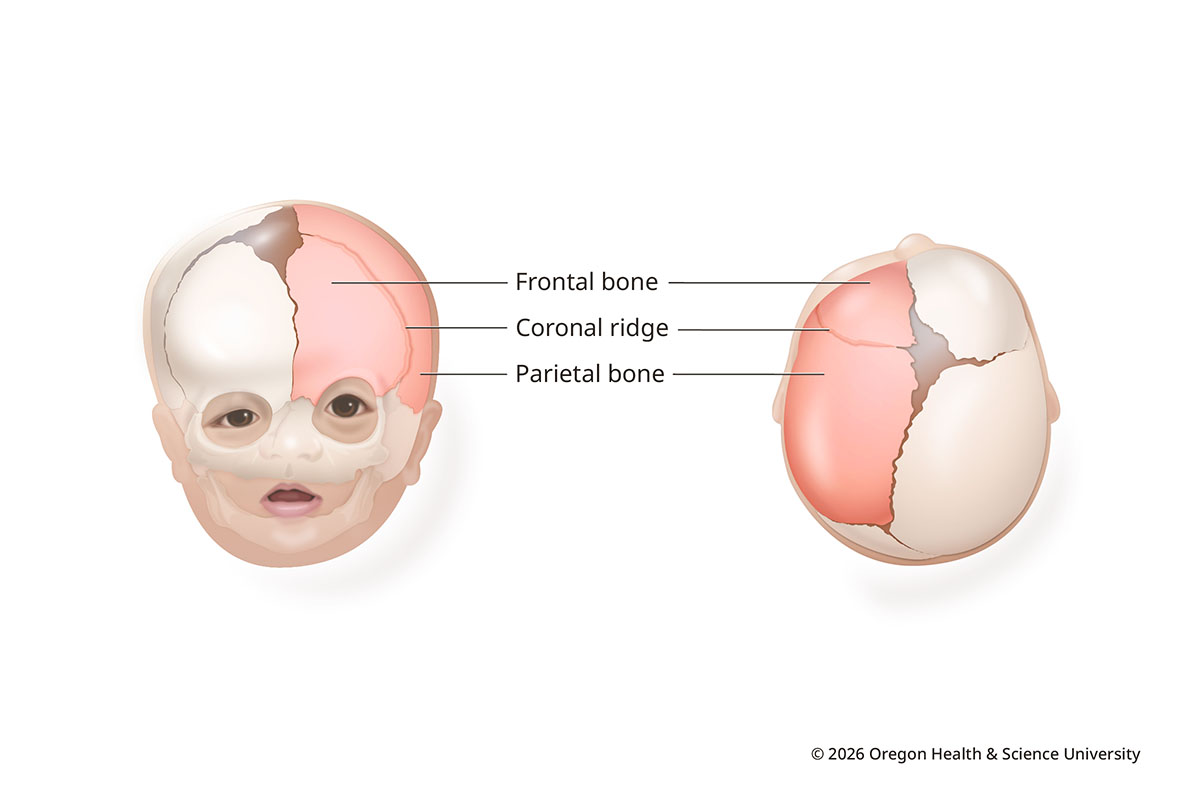
In coronal synostosis, one or both coronal sutures close early. The sutures run ear to ear across the top of the head.
When one suture closes early, the forehead is flat on the affected side and more prominent on the other. The affected eye socket rises and tilts. Coronal synostosis makes up about 20-30% of craniosynostosis cases.
Coronal Synostosis
Bilateral coronal synostosis (anterior brachycephaly)
Bilateral coronal synostosis happens when both coronal sutures close too soon. With forehead growth restricted, the skull grows upward and outward. The head can be wide ear to ear and short front to back. The forehead may be high and flat, and the head may look tall.
Bilateral Coronal Synostosis
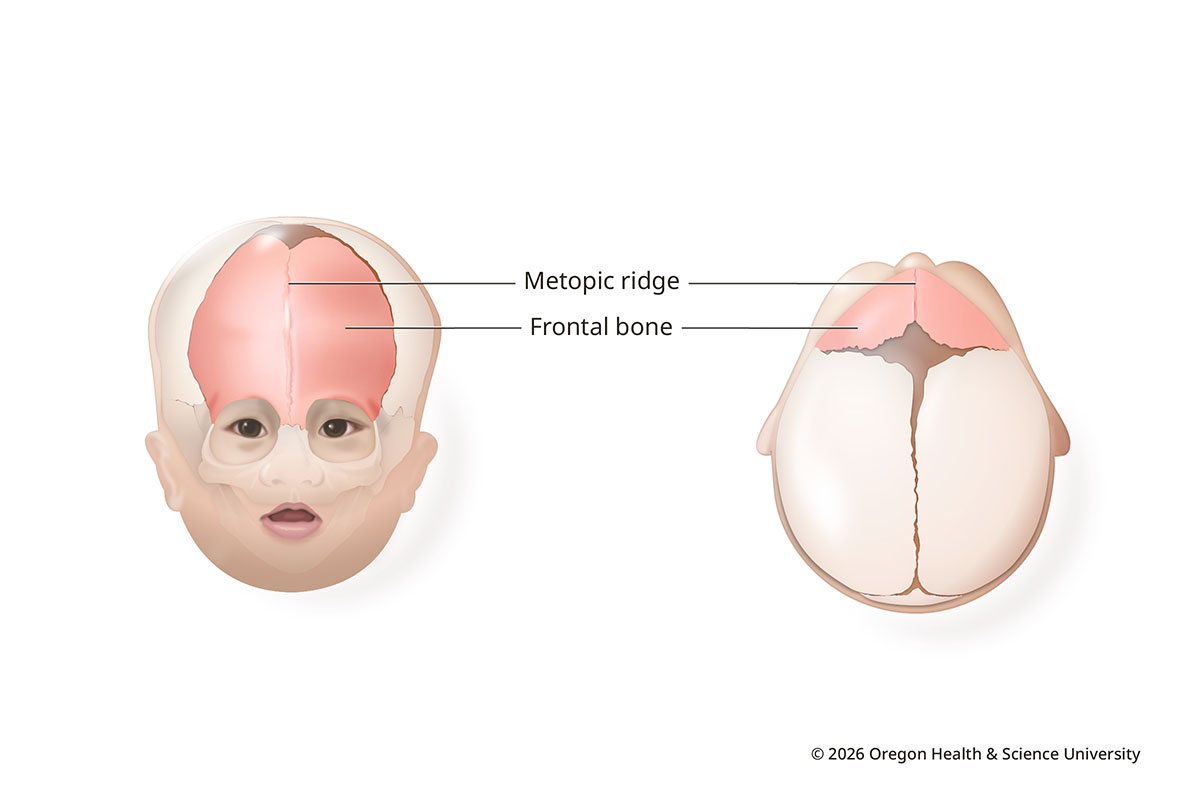
Metopic synostosis (trigonocephaly)
In metopic synostosis, the vertical seam in the middle of the forehead fuses early. This can result in a narrow, pointed forehead. Bone can form a ridge down the middle. Eyes may look closer together.
A ridge alone does not mean a baby has metopic synostosis, though. The metopic suture is the first to close, and a ridge is not uncommon. This type of craniosynostosis makes up about 10-20% of cases.
Metopic Synostosis
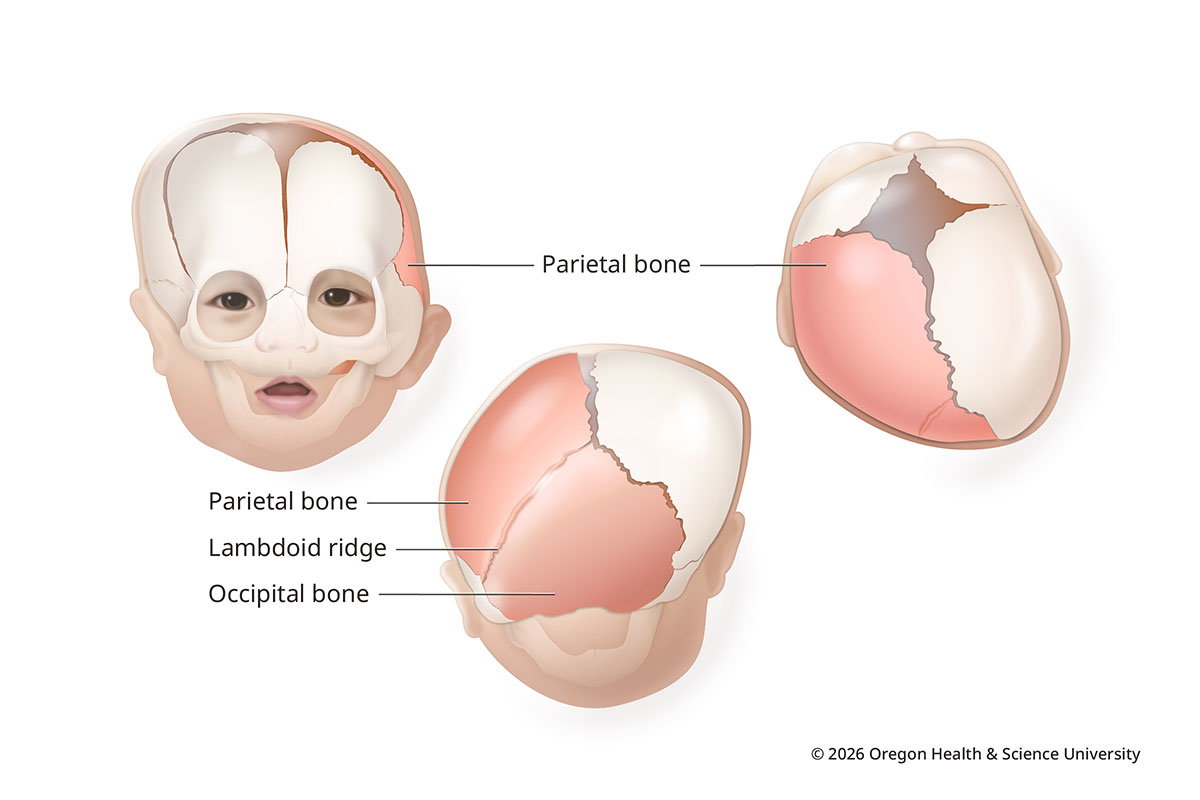
Lambdoid synostosis (posterior plagiocephaly)
Lambdoid synostosis is rare, making up less than 5% of cases. It happens when the lambdoid suture, usually on one side, closes too soon.
This can lead to flattening at the back of the head on the affected side. The skull may bulge behind the ear, and the ear may be pulled backward and downward on that side. The forehead on the other side may bulge to make space for the brain.
Lambdoid Synostosis
Syndromic craniosynostosis
In some cases, craniosynostosis is part of a genetic condition (syndromic craniosynostosis). These conditions can affect how the skull, face, hands and feet develop.
More than 150 syndromes are linked to craniosynostosis. The most common include:
- Apert
- Crouzon
- Muenke
- Pfeiffer
- Saethre-Chotzen syndromes.
Care for these conditions is more complex and may involve a team of specialists.
Learn more
- Craniosynostosis, Genetic and Rare Diseases Information Center
- Children’s Craniofacial Association
- FACES, The National Craniofacial Association
Location
OHSU Doernbecher Children’s Hospital
700 S.W. Campus Drive
Portland, OR 97239
Free valet parking for patients and visitors
For referring providers
Refer your patient to OHSU Doernbecher.
Call 503-494-8088 to:
- Seek provider-to-provider advice.
- Request education about plagiocephaly or other conditions.