Current Projects
Potassium
How the kidney controls potassium homeostasis
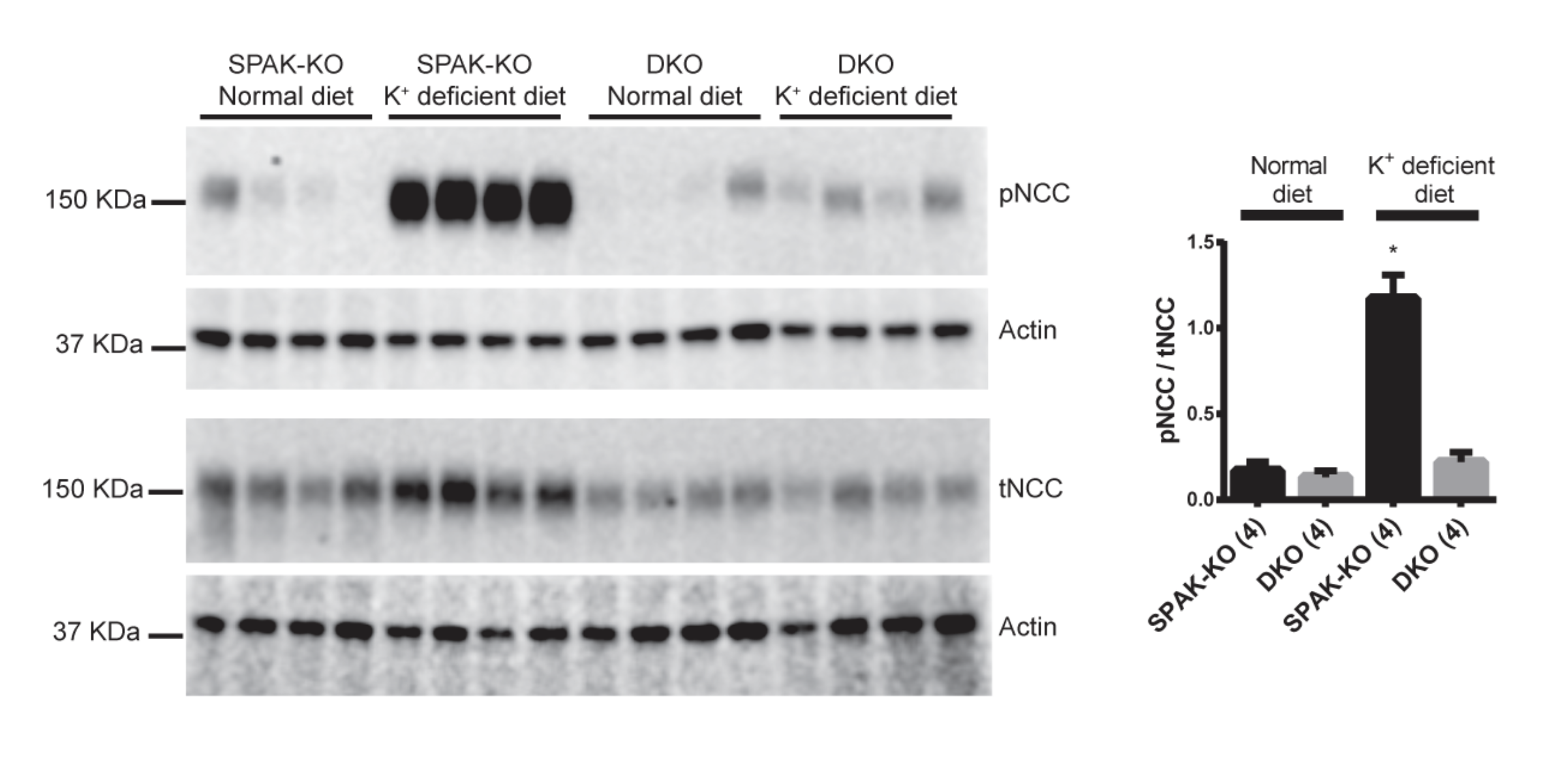
High blood potassium is another feature of the disease Familial Hyperkalemic Hypertension. Working with the Ellison lab and Wen-Hui Wang at New York Medical College, we have been working on a project to determine how the kidney senses and fine-tunes blood potassium levels. Our work has shown that a potassium channel called Kir4.1 plays a central role in this process. Kir4.1 is present in the same cells as the salt transporter NCC, which is activated by WNK kinases. When blood potassium is low, potassium exits cells which induces chloride exit. Chloride inhibits WNK kinase activity, so when there is lower chloride in the kidney cells, WNK kinases are activated. This activates NCC, which leads to effects on downstream parts of the kidney tubule that decrease potassium secretion. This then normalizes blood potassium levels. Our work involves examining how SPAK and OSR1 and Cullin Ring Ligases are involved in this process, and we are also measuring chloride concentrations in kidney tubules.
Kidney Injury
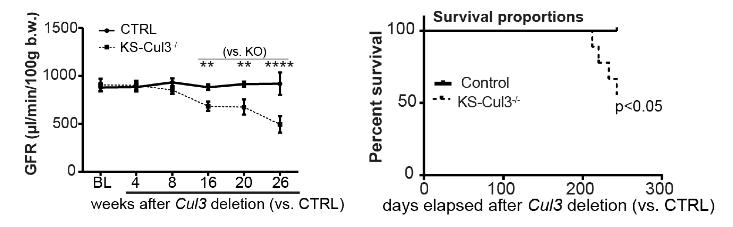
Acute injury to the kidney is frequently repaired without long-term consequences, but it can lead to the development of Chronic Kidney Disease (CKD), which manifests as electrolyte abnormalities and hypertension. CKD is a progressive disease that eventually leads to widespread renal fibrosis and renal failure. In the later stages, CKD patients require dialysis to stay alive. When we disrupted Cullin 3 in the kidney in mice (KS-CUL3-/-) they developed CKD. We are now working to determine the mechanisms by which disruption of the Cullin Ring Ligase leads to CKD. Our recent data suggest that dysregulation of the cell cycle, resulting in altered cell proliferation, may play an important role. In this project we measure renal function, and determine changes in markers of renal injury by immunofluorescence of kidney sections. We are also beginning “omics” studies to perform global analyses of changes in gene expression and abundances of different proteins to get a complete picture of the early changes occurring after Cullin 3disruption. This will allow us to identify targets of the Cullin Ring Ligase that may contribute to CKD.
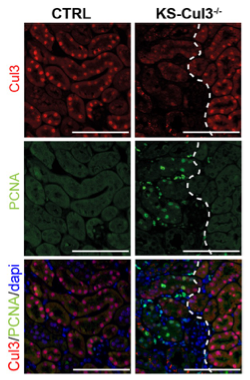
In KS-CUL3-/- mice, CUL3 is not completely disrupted as shown by red nuclear signal. In areas of the kidney lacking CUL3, we detected increased cell proliferation, as shown by PCNA expression (green).
Hypomagnesemia
Hypomagnesemia is a serum concentration of Mg2+ <1.6mg/dL and is common in hospitalized patients, with a prevalence as high as 65% in ICU patients. It can cause seizures, osteoporosis, and fatal arrhythmias; milder but still significant effects include tremors, pain, and confusion. Hypomagnesemia is also associated with worse outcomes in diabetes, cardiovascular disease, and chronic kidney disease. The distal convoluted tubule (DCT) in the kidney plays a key role in maintaining serum, with most genetic causes of hypomagnesemia affecting this kidney segment. Hypomagnesemia is very commonly seen together with hypokalemia, which may exacerbate negative outcomes. Very little is known about how hypomagnesemia affects kidney function, or how hypomagnesemia and hypokalemia interact. Our recent work explores to address this gap in knowledge. We confirmed previous work that shows that hypomagnesemia decrease NCC levels, and showed that it overrides the effect of hypokalemia to activate NCC. Current work explores mechanisms that may be involved, including changes in intracellular calcium levels and increased NCC ubiquitination.
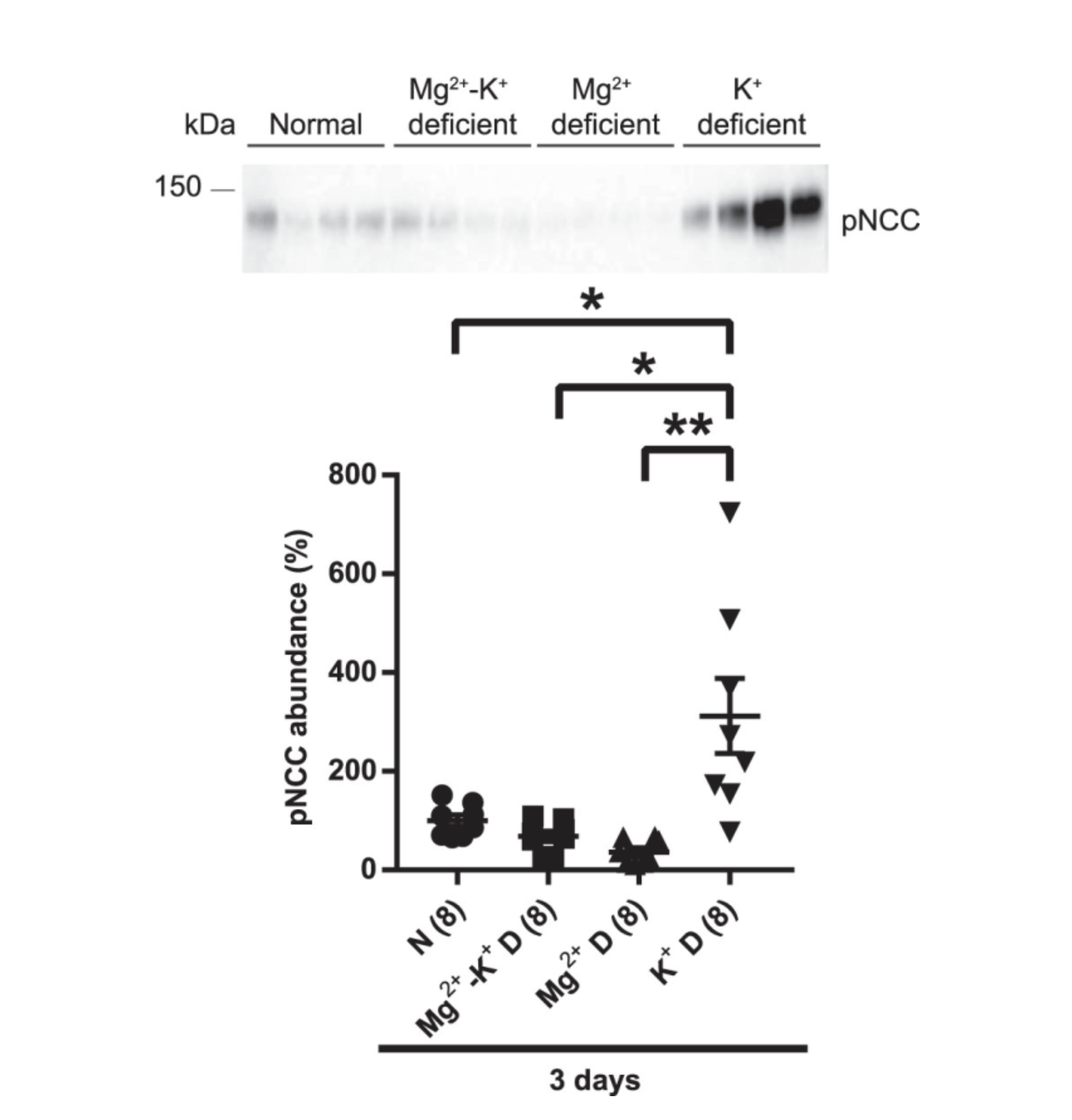
Low Mg2+ overrides the ability of low K+ to activate NCC. This might increase K+ excretion into the urine by effects on downstream kidney segments, and explain why in hospitalized patients, hypomagnesemia leads to sustained hypokalemia.
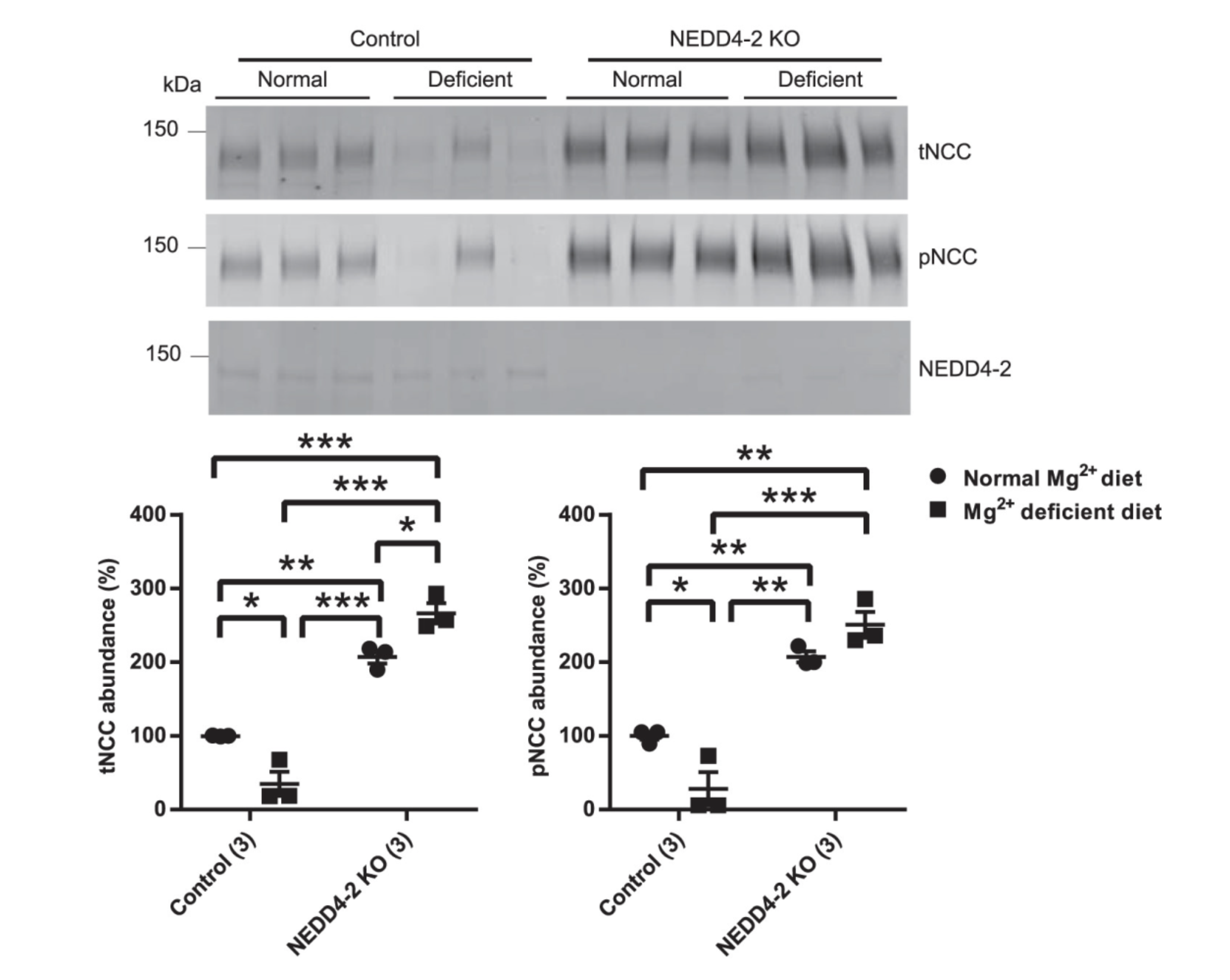
The ubiquitin ligase, NEDD4-2 tags NCC for degradation by the proteasome. We found that low Mg2+ diet did not lead to lower NCC in mice lacking NEDD4-2 in the kidney (NEDD4-2 KO), suggesting NEDD4-2 may a role in the effect of hypomagenesmia on NCC levels.