Projects
Landfear Laboratory Research
The Landfear laboratory studies the cell and molecular biology and biochemistry of single cell parasites, both Leishmania species and African trypanosomes (Trypanosoma brucei). These two related parasites cause devastating diseases, leishmaniasis (estimated 12 million people infected) and African sleeping sickness, primarily in the developing world. Because these parasites are very distant from higher eukaryotes on an evolutionary scale, they display many intriguing and unusual biological features and present fascinating systems for exploring cell and molecular biology. In addition, the current drugs for both parasites are far from optimal, and there is an urgent need to discover new leads for development of more potent and less toxic therapies. Hence, our research covers both basic biology and novel drug development for these important parasites.
Background on Leishmania and African Trypanosomes
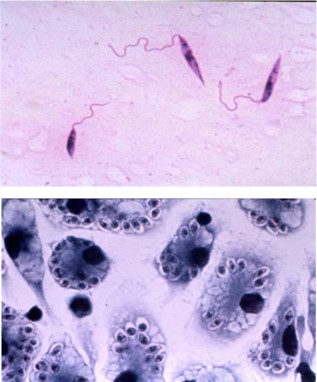
Both parasites live in the very different environments of an insect vector (sand fly for Leishmania, tsetse fly for trypanosomes) and a mammalian host, and they have evolved very different life cycle forms to colonize these two distinct environments. Leishmania parasites (Figure 1) live as extracellular promastigotes (PRO, top image) in the gut of the sand fly vector and as intracellular amastigotes (AM, bottom image) within parasitophorous vacuoles of mammalian macrophages (host immune cells). PRO have a long highly motile flagellum and can be cultured readily in various media. AM have a very short non-motile flagellum, they can be grown inside murine or human macrophages, they can infect model animals such as mice, and they can be cultured in the absence of host cells as axenic amastigotes (AxAM) by mimicking the acidic pH and increased temperature the parasites
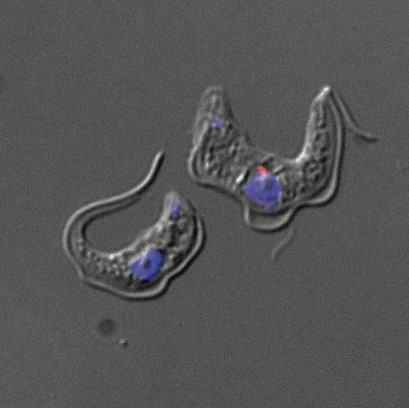
experience within the parasitophorous vacuole. There are robust molecular genetic tools to study these parasites and the functions of specific genes and proteins.
African trypanosomes live in the gut and salivary glands of tsetse fly vectors and in the bloodstream of the mammalian host (Figure 2).
Three of the major projects of the laboratory dealing with Leishmania and trypanosomes are outlined below.
Project 1: Assembly and Function of the Flagellar Membrane.
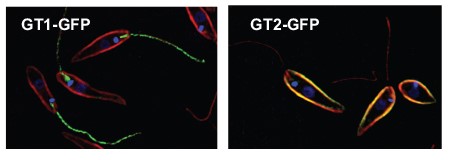
Flagella play central roles in the biology of Leishmania and African trypanosomes. They are responsible for parasite motility, but they also serve as antenna to sense the environment, as adhesive organelles to allow attachment of the parasite to both epithelia of the insect vector and the vacuolar membranes of mammalian macrophages, as secretory organelles, and as attachment sites for parasite mating. The surface membrane of the flagellum is critical for these specialized functions, so it is not surprising that the flagellar membrane is unique, containing proteins that target selectively to this membrane where they serve diverse functions. One example is shown in Figure 3. The GT1 glucose transporter (tagged with the green fluorescent protein GFP) targets specifically to the Leishmania flagellum, whereas a closely related glucose transporter GT2 is excluded from the flagellum and traffics to the membrane surrounding the cell body. GT1 appears to serve a role in sensing extracellular glucose, whereas GT2 is the principal transporter for uptake of the nutrient glucose into the parasite. Major objectives of our laboratory are to 1) dissect the molecular mechanisms these parasites use to selectively target specific proteins to the flagellar membrane where they carry out their dedicated functions, and 2) understand the functions of flagellar membrane proteins in the biology of the parasite.
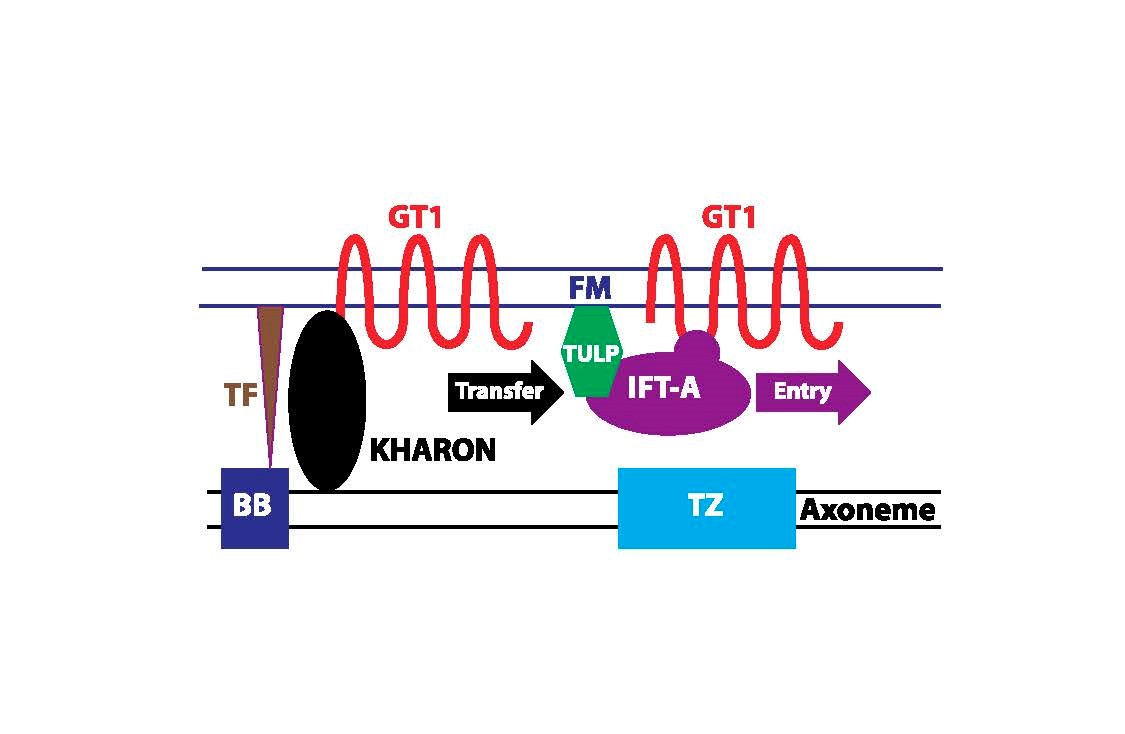
For GT1, we have identified a novel protein, called KHARON, that interacts with this transporter and is essential for its trafficking to the flagellar membrane. Studies on KHARON have demonstrated that it binds to the flagellar cytoskeleton (the tubulin-based axoneme) at the base of the flagellum and thus brings GT1 into this initial domain of the flagellum. Recent work indicates that KHARON then transfers GT1 onto Intraflagellar Transport (IFT) particles, multiprotein complexes that move GT1 into the flagellar membrane using the motor proteins, such as kinesin, that are associated with IFT particles (Figure 4). Further work on this novel flagellar trafficking mechanism is underway. .
Despite the central role of KHARON in flagellar trafficking of GT1, many other flagellar membrane proteins do not employ KHARON to enter the flagellum, and they must interact with other trafficking machinery for flagellar membrane localization. We are now studying several such proteins to identify associated partners that allow them to enter the flagellum and, in some cases, to localize to specific subdomains of the flagellar membrane. The objective is to first identify partner proteins required for flagellar trafficking and to dissect the molecular mechanisms whereby they promote flagellar localization.
Another objective is to probe the functions of diverse flagellar membrane proteins to understand how they endow the flagellum with its critical biological properties. Flagellar membrane proteins of interest include one that tethers the base of the flagellum to the cell body membrane, channels that are involved in sensing osmolarity and potentially mediating directed motility within the sand fly vector, and components of the cAMP signal transduction machinery that are probably important both for colonization of the insect and for successful infection of host macrophages.
Relevant Publications:
Tran, K.D., Rodriguez-Contreras, D., Vieira, D.P., Yates, P.A., David, L., Beatty, W., Elferich, J., and Landfear, S.M. KHARON1 mediates flagellar targeting of a glucose transporter in Leishmania mexicana and is critical for viability of infectious intracellular amastigotes. J. Biol. Chem. 288:22721-22733 (2013)
Sanchez, M., Tran, K.D., Valli, J., Hobbs, S., Johnson, E., Gluenz, E., and Landfear, S.M. KHARON is an essential cytoskeletal protein involved in the trafficking of flagellar membrane proteins and cell division in African trypanosomes. J. Biol. Chem., J. Biol. Chem. 291:19760-19773 (2016).
Kelly, F.D., Sanchez, M.A., and Landfear, S.M. Touching the surface: Diverse roles for the flagellar membrane in kinetoplastid parasites. Microbiol. Mol. Biol. Rev. 84:e00079-19 (2020).
Kelly, F.D., Tran, K.D., Hatfield, J., Schmidt, K. Sanchez, M.A., and Landfear, S.M. A cytoskeletal protein complex is essential for division of intracellular amastigotes of Leishmania mexicana. J. Biol. Chem. 295:13106-13122 (2020).
Landfear, S.M. New vistas in the biology of the flagellum – Leishmania parasites. Pathogens 11:447 (2022).
Project 2: Essential Functions of the Neddylation Pathway in Leishmania Parasites
A major discovery in biology was that the small (76 amino acid) protein ubiquitin could be covalently attached to specific lysine residues on many cellular proteins and could thus modify their activities. A principal, albeit not exclusive, function of ubiquitination is to target the modified proteins for proteolytic degradation, thus determining which proteins are stable and which are removed from cells under various physiological conditions. Intriguingly, the ubiquitin modification pathway itself is subject to regulation by a process in which another ubiquitin-related protein, NEDD8, is covalently attached to one subunit (the cullin protein) of the so-called E3 ubiquitin ligase enzymes that attach ubiquitin to its substrate proteins. Once NEDD8 is attached to a cullin, the E3 enzyme is activated to ubiquitinate its substrates, and thus neddylation (the covalent attachment of NEDD8 to proteins) ultimately controls the composition of the cellular proteome. Hence, a major area of study is the neddylation pathway, which itself contains 3 enzymes, E1, E2, and E3, that activate NEDD8 and ultimately attach it to cullins (Figure 5). Neddylation is of major biomedical importance, as overactive neddylation is a dysregulation associated with many cancers, and inhibiting the overactive neddylation pathway is being pursued actively for anti-cancer therapies.
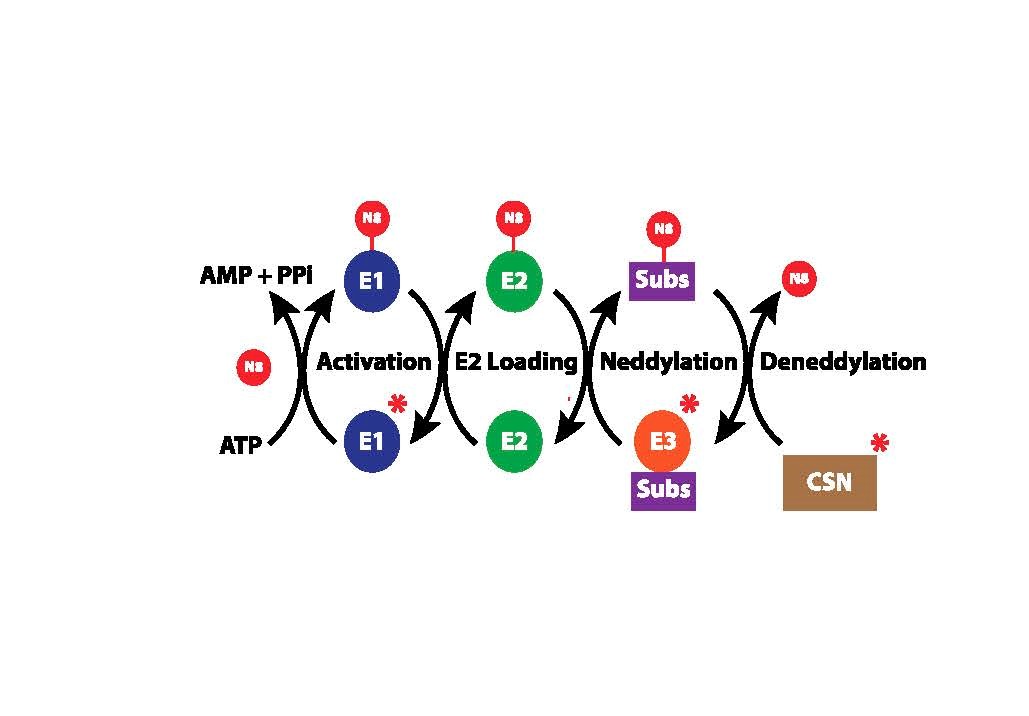
Our laboratory has established a collaboration with the laboratories of Drs. Kip Guy (University of Kentucky, chemical biology) and Brenda Schulman (St. Jude Cancer Research Hospital, structural biology), two prominent researchers on neddylation in mammalian cells. The objective of this collaborative project is to both understand the basic biochemistry and biology of the neddylation pathway in Leishmania parasites and to target it for development of novel anti-parasitic drugs. Our laboratory has demonstrated, using conditional gene knockout strategies, that NEDD8 and the E1 and E3 enzymes of this pathway are essential for parasite viability and that E1 and E3 can thus be targeted for therapeutic intervention. In collaboration with the Guy and Schulman laboratories, compounds are now being developed, by structure guided drug design, that can selectively inhibit the parasite, but not the human, neddylation enzymes and that could be developed as new anti-parasitic drugs. Such inhibitors of neddylation can also be employed to explore the function of protein neddylation in Leishmania parasites.
Relevant Publications:
Scott, D.C. et al. Blocking an N-terminal acetylation-dependent protein interaction inhibits an E3 ligase. Nature Chemical Biology 13:850-857 (2017)
Project 3: Using Phenotypic Screening to Discover Novel Compounds for Anti-leishmanial Drug Development
One approach to drug discovery is to target a specific biological pathway that has been demonstrated to be essential for the parasite, as in Project 2 above. An alternative approach is to carry out large-scale screens of compound libraries to identify small molecules with potent activity against the parasite, a method referred to as ‘phenotypic screening’. The initial ‘hits’ from a phenotypic screen can then be explored for those with the greatest potential for drug development.
We have engaged in two separate collaborative projects that initiated with such phenotypic screens. The first project is a collaboration with the laboratory of Dr. Kip Guy, mentioned in Project 2, and the second is a collaboration with Dr. Jane Kelly (Portland State University, pharmaceutical chemistry). Intriguingly, the collaboration with the Guy laboratory led to discovery of two completely different small molecules that inhibit the same critical biosynthetic pathway in Leishmania parasites. Both compounds cause extensive changes in the parasite metabolome, demonstrating the core importance of this pathway for parasite metabolism. This discovery involved yet another collaboration with Dr. Mike Barrett (University of Glasgow, metabolomics) to identify the pathway that is targeted by these two compounds. Efforts are now underway to both explore the biochemical pathway and to optimize the two initial lead compounds for drug development. Another collaboration with Dr. Kelly’s laboratory has exploited a group of structurally related natural product derived compounds that have demonstrated efficacy against malaria parasites. Many of these compounds show potent activity against Leishmania parasites, and we are now exploring improved derivatives of the initial hits to identify analogs that have efficacy against Leishmania parasites in a murine model of infection. Ultimately, we plan to identify the target or targets of these compounds to allow both the development of novel anti-parasitic drugs and an exploration of the relevant pathway.
Relevant Publications:
Ortiz, D., Guiguemde, W.A., Hammill, J.T., Carrillo. A.K., Chen, Y., Connelly, M., Stalheim, K., Elya, C., Johnson, A., Min, J., Shelat, A., Smithson, D.C., Yang, L., Zhu, F., Guy, R.K., Landfear, S.M. Discovery of novel, orally bioavailable, antileishmanial compounds using phenotypic screening. PLoS Negl. Trop. Dis. 11:e0006157 (2017).
Hammill, J.T., Sviripa, V.M., Kril, L.M., Ortiz, D., Fargo, C.M., Kim, H.S., Chen, Y., Rector, J., Rice, A.L., Domagalska, M.A., Begley, K.L., Liu, C., Rangnekar, V.M., Dujardin, J.-C., Watt, D.S., Landfear, S.M., and Guy, R.K. Amino-substituted 3‑aryl- and 3‑heteroarylquinolines as potential antileishmanial agents. J. Med. Chem. 64:12152-12162 (2021).
Kancharla, P., Li, Y., Yeluguri, M. Dodean, R.A., Reynolds, K.A., and Kelly, J.X. Total synthesis and antimalarial activity of 2-(p-hydroxybenzyl)-prodigiosins, isoheptylprodigiosin, and geometric isomers of tambjamine MYP1 isolated from marine bacteria. J. Med. Chem. 64:8739-8754 (2021)
Kancharla , P. Ortiz, D., Fargo, C.M., …, Kelly, J.X., and Landfear, S.M. Discovery and optimization of tambjamines as a novel class of antileishmanial agents. J. Med. Chem. 67:8323-8345 (2024)