Projects
Overview
Our lab focuses on two human herpesviruses: herpes simplex virus (HSV) and human cytomegalovirus (HCMV). We study interactions between these viruses and host cells, the immune system and viral pathogenesis. One major focus is on how HSV moves within and between neurons. HSV establishes latency in neurons and periodically reactivates, then spreads along neuronal axons to peripheral tissues. HSV infections in the cornea can lead to scarring and blindness - HSV is still the major infectious cause of blindness in the western world. We have identified two HSV proteins that promote transport of HSV on microtubules motors in neuronal axons (Polcicova, K. et al. 2005, PNAS 102:11462). Other studies involve how HSV crosses the nuclear envelope (Farnsworth et al. 2007. PNAS 104: 10187-92). Our work on HCMV has focused on viral immune evasion and inhibition of the MHC class I and II antigen presentation pathways. We have studied the molecular mechanisms involved in the inhibition, or destruction, of MHC class I or II proteins by viral proteins (Hegde et al. J. Exp. Med. 2005 202:1109 and 2006. J. Biol. Chem. 281:20910. Other studies on HCMV involve virus tropism, how HCMV infects different cells by using different receptors (Ryckman, B et al. 2006, J. Virol. 80:710 and 2007 J. Virol in press).
In detail
Herpes simplex virus cell-to-cell spread and ocular disease.
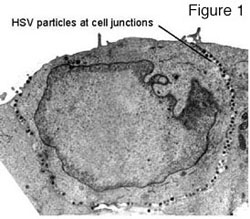
HSV has studied epithelial and neuronal cell junctions for many millions of years in order to spread through mucosal, ocular and neuronal tissues. These viruses move swiftly and efficiently between infected and uninfected cells by cannibalizing the cell’s intracellular sorting machinery to direct viral progeny to specific cell surfaces i.e. epithelial and synaptic junctions (reviewed in Johnson and Huber 2002). For example, several viral membrane proteins, e.g. gE/gI and gB, bind clathrin adaptor proteins, acidic cluster binding proteins PACS-1 and PACS-2, as well as other components of the trans-Golgi network (TGN)/endosomal sorting machinery. These interactions determine movement of progeny viruses (produced in the nucleus) to cell junctions. We have shown that nascent HSV particles are specifically directed to epithelial cell junctions and are not found on apical surfaces but instead accumulate in the spaces between cultured epithelial cells (see figure one and Johnson et al. 2001). We are attempting to define intracellular events and cellular components that participate in this process. A model explaining how sorting in the TGN can promote cell-to-cell spread of HSV is shown in figure two. HSV glycoproteins, gE and gI form a heterodimer known as gE/gI and this complex localizes to the TGN by virtue of numerous sorting domains in the cytoplasmic domains of these proteins.
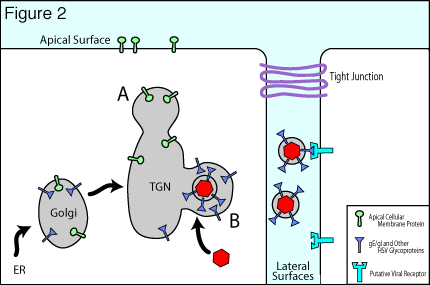
This promotes virus envelopment in the TGN, and specifically in subcompartments of the TGN denoted B in figure two. Virus particles that are sorted into compartments B are, in turn, directed to epithelial cell junctions by mechanisms used by the cell to sort cellular proteins to basolateral surfaces in polarized cells. Other cellular components are sorted to apical cell surfaces (A in figure two) by virtue of sub-compartmentalization into other TGN compartments. In general, the molecular details of apical versus basolateral sorting in polarized cells are poorly understood and we believe that our studies should add to this understanding. By acting to move nascent virions to cell junctions, gE/gI can speed spread of virus between closely packed cells and produce directed spread. Moreover, gE/gI also binds cellular receptors that are concentrated at cell junctions, thereby promoting virus movement into adjacent cells.
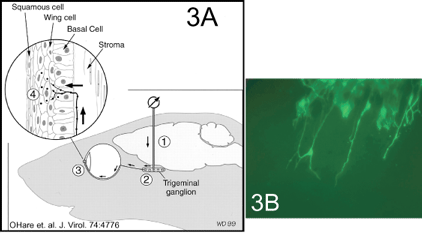
Sorting of HSV proteins and directed transport of virus particles also occurs in neurons. Virus infecting neurons at axon tips move to the nucleus along microtubules. Later (often after latent virus reactivates) viral progeny are specifically delivered down axons via other microtubule motors, and appear to be sorted away from dendrites. We are studying HSV transport within sensory neurons following infection in the cornea. Moreover, we can directly inject HSV into the trigeminal (TG) ganglia and characterize virus movement back to the cornea (figure three A). In many cases, we track viruses by using recombinants expressing GFP or fusion proteins in which GFP or RFP is fused to a viral membrane protein (figure three B: See green axons of neurons infected in the TG ganglia and projecting into the cornea).
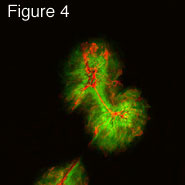
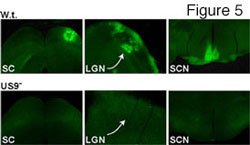
In recent studies have we identified and characterized an HSV membrane protein, US9, that functions to promote HSV cell-to-cell spread. Unlike gE/gI, US9 functions solely in neurons. A US9- mutant was able to spread normally in the cornea, moving to hundreds of other epithelial cells by 2 days post-infection (see figure four, green is GFP expressed by a recombinant HSV and red is staining with anti-HSV antibodies). Spread of the US9- virus was identical to wild type HSV-1 (not shown). However, US9 could not spread from infected neurons to other cells in the nervous system. In figure five, viruses were injected into the retina, and spread from neuron cell bodies in the retina to retinorecipient regions of the brain was examined. The US9- mutant did not spread to any of the regions of the brain (the SC, the LGN and SCN) that are connected to the retina. A control wild type virus (w.t.) expressing GFP spread to all three regions. We are attempting to characterize the molecular mechanisms by which US9 promote this virus movement in neurons.
US9 apparently contains sequences that couple HSV structural proteins onto microtubule motors causing these viral building blocks to move down axons. Exiting cells, HSV assembles, not in the cell body, but at axon tips. We are also using the US9- mutant and other mutants to study HSV keratitis. One particularly interesting question relates to the origin of HSV antigens that cause this inflammatory disease in the cornea. There is clearly persistent HSV in the cornea and this could trigger immune responses that ultimately damage the cornea. Alternatively, infectious HSV that reactivates in latently-infected neurons could reinfect the cornea producing immunity that causes corneal scarring. Since the US9- mutant cannot return to the cornea from infected neurons, we can test which of these two possibilities is correct using mouse models.
Herpesvirus immune evasion.
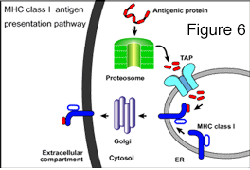
The second major focus of our research involves recognition of viruses by the host’s immune system and evasion of this immunity. In order to persist or remain latent for decades, herpesviruses have evolved many carefully crafted mechanisms to modulate immune responses or avoid recognition. Some years ago, we identified the first herpesvirus protein that blocks detection of infected cells by CD8+ T lymphocytes. HSV ICP47 inhibits TAP, the Transporter associated with Antigen Presentation, a cellular protein that pumps antigenic peptides into the ER where they are loaded onto MHC class I proteins (figure six). Class I proteins move from the ER through the Golgi apparatus to cell surface and present antigenic peptides to the T cell receptors of T lymphocytes. By blocking TAP, ICP47 inhibits recognition of HSV-infected cells by T cells. Since those studies, a large number of viral inhibitors of TAP and other steps in the MHC class I antigen presentation have been described. More recently, we have probed HCMV for other, novel immune evasion mechanisms. HCMV has about 2.5 times the coding capacity of HSV, so that there is the potential to express a huge number of immunomodulatory molecules. Moreover, HCMV replicates very slowly, increasing vulnerability to host immune responses. To deal with the immune system HCMV expresses an impressive array of immune evasion and immunomodulatory proteins. Moreover, HCMV infects a relatively large number of cell types in the body, including several that express MHC class II proteins. We became interested in whether HCMV might block, not only the MHC class I pathway, but also the MHC class II antigen presentation pathway.
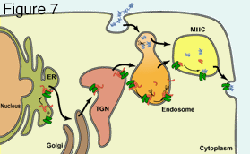
Normally, MHC class II functions in “professional” antigen presenting cells to present exogenous antigens to CD4+ T lymphocytes. By exogenous, we mean extracellular antigens that are taken up by endocytosis or phagocytosis (refer to figure seven). Class II proteins are synthesized in the ER as a complex of alpha, beta and invariant (Ii) chains that move to the Golgi apparatus and on to MHC class II loading compartments that are similar to lysosomes. In these acidic loading compartments, class II proteins are loaded with peptides that are derived from proteins taken up into endosomes, and degraded by acidic proteases. We recently identified two HCMV proteins that inhibit the class II pathway in different ways. HCMV US2 causes class II proteins present in the ER to be retrotranslocated out of the ER and into the cytoplasm where the proteins are degraded by proteasomes. HCMV US3 reduces binding of the invariant chain (Ii) that normally stabilizes class II alpha/beta complexes in the ER and sorts complexes to class II loading compartments. Without Ii, class II complexes are mislocalized in cells and do not reach loading compartments. These are two interesting examples of how HCMV can reduce recognition by CD4+ T cells, in addition to evading CD8+ T cells.

By reducing class I and II proteins on the cell surface, viruses expose themselves to natural killer (NK) cells that recognize cells with reduced MHC proteins. To combat NK cells, HCMV expresses an MHC class I homologue, UL18, that apparently replaces the lost surface class I. Recently, we characterized a second HCMV protein, UL16, that reduces exposure to another NK recognition receptor, NK2G-D. NK2G-D recognizes MICA and MICB which are MHC class I proteins (see figure eight). Normally, HCMV reduces surface MHC class I by inhibiting the class I pathway, but also triggers high level transcription of the MICA and MICB genes that are regulated by heat shock promoters. The appearance of MICA and MICB on the surfaces alerts NK and T cells with NK2GD receptors that HCMV is present. These lymphocytes become activated and would kill the HCMV-infected cells, unless HCMV counters this. We found that UL16 binds MICB and mislocalizes the cellular protein, reducing recognition by NK cells (Dunn et al 2003). Our continuing studies involve mapping and identification of other novel viral inhibitors of the immune system and detailed characterization of how these viral proteins function. The work involves the cell biology of antigen presentation pathways, use of recombinant virus expression vectors, and characterization of T and NK cell recognition mechanisms.
Regulation of herpesvirus by cellular immunity and HSV corneal keratitis.
We are also interested in the role of CD8+ and CD4+ T lymphocyte responses in controlling HSV latency and triggering keratitis. There is evidence that anti-HSV T lymphocytes interact with HSV-infected neurons in a specific but yet benign manner. This is unlike the normal interaction between cytotoxic CD8+ T cells and cells they kill. Instead, expression of viral proteins and replication in neurons is curtailed but without harm to the neurons so that latency is established or maintained. Apparently, these T cells produce molecules e.g. cytokines, that dampen virus replication. In collaboration with others, we are beginning to dissect the HSV molecules that are recognized by these T cells and to understand how the T cells interact with neurons in this novel manner.
T cells can be beneficial in suppressing virus replication in neurons, however these cells can also cause major disease in the cornea. CD4+ and CD8+ T lymphocytes that recognize viral antigens in the eye are thought to be the major causes of inflammation that damages the cornea in keratitis. We are attempting to identify the T cell antigens and to determine how these antigens reach the cornea. One perplexing aspect of keratitis involves observations that there are very low or undetectable levels of HSV antigens present in the stroma (the deeper layer of the cornea) after HSV reactivation. One interesting possibility is that HSV membrane glycoproteins move very rapidly and in a directed fashion along axon microtubules from sensory neuron cell bodies in the ganglia to the stroma and these proteins then act as T cell antigens to promote inflammation. Indeed, there is evidence that mice infected with an HSV recombinant expressing a viral glycoprotein fused to GFP displays fluorescent signal transferred from ganglia into the cornea and long before virus appears in the cornea. Therefore, we are characterizing traffic of viral membrane proteins from the ganglia to the cornea by using fluorescent-tagged glycoproteins as described above. In addition, we are using T cell clones that recognize epitopes in the glycoprotein fusion proteins to characterize T cell recognition and traffic in HSV keratitis.