Projects
Endothelial cells
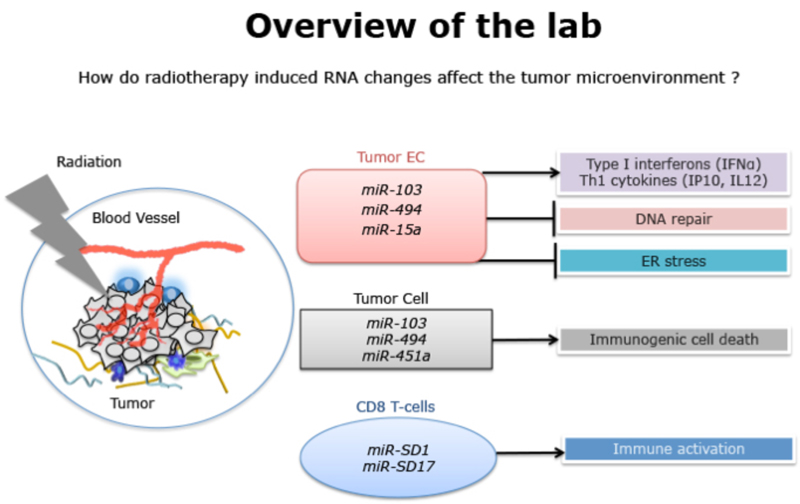
To build on our observation that miRs had a fundamental role in DNA repair, we screened for miRs that were dysregulated with different DNA damaging agents. We identified a novel, interrelated signature of microRNAs (miR) in ECs that inhibit angiogenesis by downregulating a network of DNA repair proteins. The top miR candidate in our signature, miR-103 inhibits angiogenesis in vitro and in multiple tumor models in vivo. We characterized the mammalian three prime exonuclease TREX1 as a major target of miR-103. miR-103 mediated TREX1 silencing provokes a potent inflammatory cytokine profile, upregulation of positive immune costimulatory molecules, decrease of immunosuppressive macrophage and induction of cell death pathways on mouse tumors. These observations highlight that targeting TREX1 reprograms a tumor permissive immune microenvironment to a tumor suppressive immune microenvironment. We have initiated a small molecule drug discovery approach to inhibit TREX1. We designed a high-throughput screening compatible, fluorescence based strand displacement assay to identify inhibitors of TREX1 enzymatic activity. We have established a collaboration with the Sanford-Burnham-Prebys Institute in La Jolla to use this assay for the discovery of agents that will disrupt TREX1 to provoke anti-tumor immune responses.
We are now broadly investigating:
- How does DNA damage induce these miRs in ECs?
- How does the DNA repair machinery drive or influence angiogenesis?
- What are the functional consequences and potential therapeutic utility of disrupting this specific DNA repair network in vivo in experimental models of cancer and vascular hyperproliferation?
Our proposed studies will elucidate the mechanistic basis for the cross talk between angiogenic growth factor signaling and the DNA damage repair pathway. Importantly, these studies open a new avenue for understanding the biological responses of ECs to DNA damage. Answering these questions will provide us with a framework for the development of novel anti-angiogenic agents that target this network.
Immune cells
Similarly, although the effects of radiation on tumor cells and the vasculature are well documented, the impact of radiation on CD8 T-cell function is not completely understood. We have now identified specific miRs that are dysregulated in human and mouse CD8 T-cells in response to radiation. We are evaluating the function of these miRs in the context of the tumor microenvironment and monitoring their utility as surrogate biomarkers of immune function.
MicroRNA guided ‘radiogenetics’ for T-cell engineering
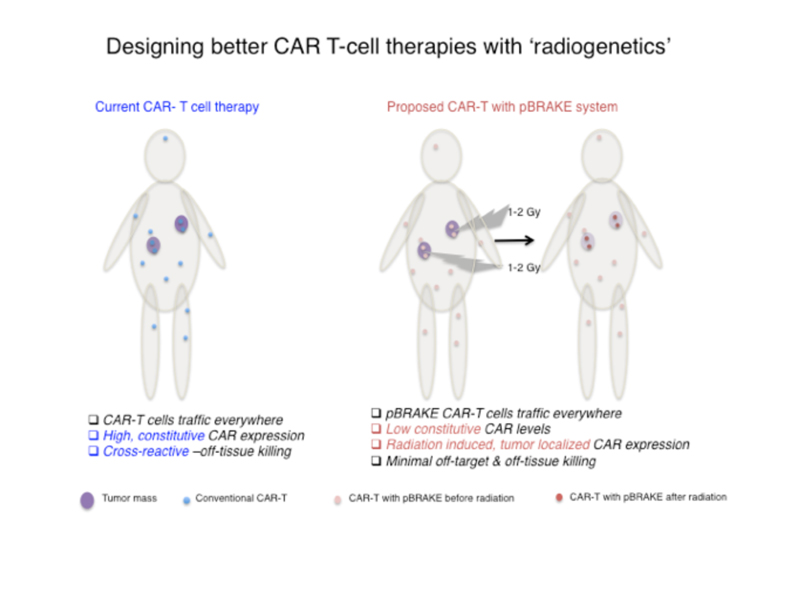
Genetically engineered T-cells as therapeutic agents offer an unprecedented opportunity for personalized cancer treatment and circumvent some of the challenges posed by tumor heterogeneity. There are currently 118 CAR-T cell therapy clinical trials targeting a number of cancers. However, there are also emerging challenges in the safety of these treatments. For instance, hyper immune activation and off-tissue reactivity such as cerebral edema or neurotoxicity have been reported in CAR-T infused patients. Moreover, the current third generation CAR T-cells have been shown to persist for up to four years, making self-reactivity and autoimmune disease in other non-tumor tissues a significant concern over time. Therefore, a strategy that would minimize off-tissue activation is anticipated to significantly improve the safety and utility of CAR-T cells.
We are designing a novel approach to minimize off-tissue activation of CAR-T cells by controlling the timing and localized expression of the CAR protein via radiation. Taking advantage of mechanisms employed by miRs, we have designed synthetic 3’ untranslated regions (UTRs) that can restrict CAR expression by binding to endogenous CD8 T-cell miRs. We have also identified a subset of these CD8 T-cell miRs that are downregulated by radiation. Synthetic UTRs that harbor binding sites for these radiation suppressed miRs act as a switch that turns ON gene expression in response to radiation. Therefore, we are designing fourth generation CAR plasmids with synthetic UTRs (pBRAKE) that stay in the OFF state until localized radiation treatment. This approach would enable external, radiation guided spatial and temporal induction of CAR expression on CD8 T-cells restricting their activation to the field of radiation.