Unni Lab

Vivek K. Unni
MD, PhD, Columbia University 2003
Faculty Profile
unni@ohsu.edu
503-494-7230
Vivek joined the Department of Neurology and the OHSU Parkinson Center in September 2011. He is now an Associate Professor, Director of the Jungers Center for Neurosciences Research, and the John Hammerstad, MD, Professor of Basic Research of Movement Disorders at OHSU. Vivek received a BS in Chemistry and Biological Sciences from Stanford in 1995. He then earned a MD and PhD from Columbia University in 2003, where his research in Steven Siegelbaum’s lab was on synaptic plasticity in the hippocampus, using multiphoton imaging and whole-cell patch clamp recording methods. After a medical internship at St. Luke’s-Roosevelt Hospital in New York City, he completed a neurology residency and movement disorders fellowship at Massachusetts General Hospital/Brigham & Women’s Hospital. Before joining OHSU, Vivek was an Instructor in Neurology at Harvard Medical School and a research fellow in the laboratories of Bradley Hyman and Pamela McLean at Mass General. During his fellowship, he pioneered the development of approaches to study alpha-synuclein in the living brain of mice using in vivo multiphoton imaging through “cranial windows.”
In his recent work, Vivek is using in vivo multiphoton imaging to examine the aggregation of alpha-synuclein in mouse models of Parkinson’s Disease (PD) and Lewy Body Dementia (LBD), and a variety of techniques to study the novel hypothesis, developed in his lab, that alpha-synuclein plays unexpected roles in the cell nucleus in DNA repair. Below is a 5 minute video by Vivek, produced by the World Parkinson Congress for their 2023 Barcelona meeting, describing what is known about the normal function of alpha-synuclein.
Research
The Role of Protein Aggregation in Neurologic Disease
Although many neurodegenerative diseases are characterized by the abnormal aggregation and accumulation of specific proteins, the exact protein in each disease varies (e.g. alpha-synuclein in PD and LBD, beta-amyloid and tau in Alzheimer’s Disease, tau and TDP-43 in forms of Fronto-temporal Dementia and Amyotrophic Lateral Sclerosis, huntingtin in Huntington’s Disease, etc.). Of all these diseases, the best evidence that simply increasing the specific protein level can cause disease exists for PD and LBD. One strong piece of evidence for this is that duplication (or triplication) of the SNCA gene locus coding for alpha-synuclein, which increases alpha-synuclein levels by the seemingly modest amount of 50% (or 100%), causes patients to develop PD or LBD.
The ability to test hypotheses about how alpha-synuclein contributes to this neurodegeneration in living brain could benefit greatly from the development of new experimental strategies to visualize alpha-synuclein in vivo, including its function, metabolism and aggregation within neurons and glia. For these reasons, we have spent a significant amount of effort to develop new approaches to try and tackle these problems. Our work allows us (for the first time) to visualize alpha-synuclein in the living brain in mouse models of PD and LBD using in vivo multiphoton fluorescence microscopy through cranial windows. One goal of our work is to use these mouse models of PD and LBD, where alpha-synuclein is over-expressed, to understand in the living brain how increasing levels of this protein leads to its aggregation and subsequent neuronal dysfunction and cell death.
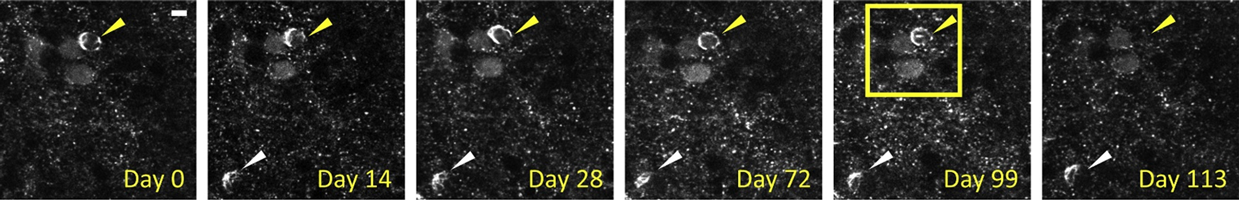
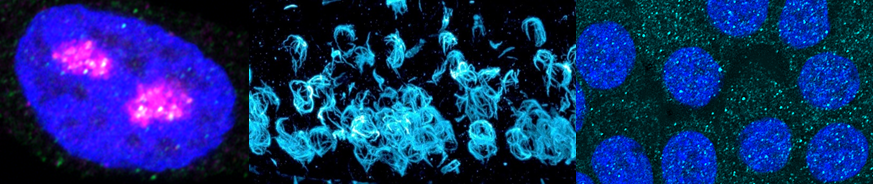
Our data demonstrate that we can visualize human alpha-synuclein fused to Green Fluorescent Protein (Syn-GFP) in the cortex of transgenic mice with cellular and synaptic resolution using this cranial window-based approach. Furthermore, we can follow individually labeled neurons and presynaptic terminals serially over a period of many months. We have paired this approach with alpha-synuclein preformed fibril seeding models to be able to image, for the first time, the formation of Lewy bodies in the living brain. Our work suggests that once a neuron forms a Lewy body it is destined to die within weeks to months, while nearby cells without Lewy bodies almost never die within this time frame. We are currently building on these strategies to test the hypothesis that alpha-synuclein may interact in vivo with other proteins relevant to neurodegeneration, like beta-amyloid and tau, and that these interactions are important for each protein’s ability to aggregate and contribute to disease.
Unexpected Roles for Synuclein in DNA Repair
Our in vivo imaging studies of alpha-synuclein aggregation in mouse models led us to postulate a new hypothesis for alpha-synuclein function in the cell nucleus in DNA double-strand break (DSB) repair. Although alpha-synuclein has been described as having a normal functional role in both the synapse and nucleus since its discovery (its name is a contraction of these two words), its function in the nucleus has been much less studied. Our observation that when a neuron forms a Lewy body it loses expression of soluble alpha-synuclein from the nucleus led us to consider whether a loss-of-function of alpha-synuclein could play a role in neurodegeneration. This work was inspired, in part, by work done by others in the pediatric cerebellar disorder Ataxia Telangiectasia, where deficiency of DSB repair is associated with Lewy body formation.
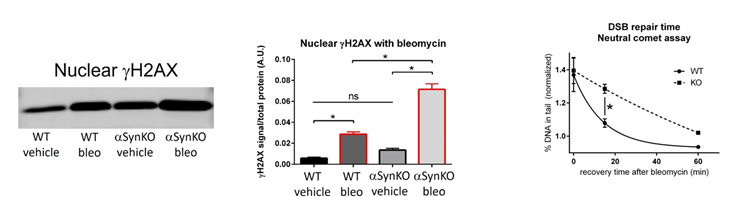
Our data demonstrate that alpha-synuclein colocalizes with multiple established DSB repair components within discrete nuclear foci in human cells and mouse brain. Alpha-synuclein knockout increases DSBs after treatment with the chemotherapeutic bleomycin and reduces DSB repair kinetics in human cells. Alpha-synuclein knockout mice show increased DSBs and this is rescued by transgenic reintroduction of human alpha-synuclein. Using a new, in vivo imaging approach that we developed, we find that synuclein is rapidly recruited to DNA damage sites in living mouse brain. Interestingly, Lewy body-bearing neurons in mouse model and patient tissue demonstrate increased DSBs. These data led us to propose a new function for alpha-synuclein in regulating DSB repair. We are currently advancing this work by studying the mechanism by which alpha-synuclein modulates DSB repair and directly testing its relevance for DSB repair in neurons.
Although the risk of all cancers is generally reduced in patients with neurodegenerative diseases, there are a few notable exceptions. The best described exception is the increased risk of melanoma in PD patients. Although the cause for this increased risk is currently unclear, melanoma does highly overexpress the same alpha-synuclein protein that aggregates in PD. Another goal of our lab is to test whether our recent discovery that alpha-synuclein is important for modulating DSB repair could provide useful insights into the mechanism behind this PD-melanoma association.
Publications
Selected publications
Boutros SW, Zimmerman B, Nagy SC, Unni VK, Raber J. (2023). Age, sex, and apolipoprotein E isoform alter contextual fear learning, neuronal activation, and baseline DNA damage in the hippocampus. Molecular Psychiatry. Feb 2. doi: 10.1038/s41380-023-01966-8.
Boutros SW, Kessler K, Unni VK, Raber J. (2022). Infusion of etoposide in the CA1 disrupts hippocampal immediate early gene expression and hippocampus-dependent learning. Scientific Reports. Jul 27;12(1):12834.
Boutros SW, Krenik D, Holden S., Unni VK, Raber J. (2022). Common cancer treatments targeting DNA double strand breaks affect long-term memory and relate to immediate early gene expression in a sex-dependent manner. Oncotarget. Jan 24;13:198-213.
Dent SE, King DP, Osterberg VR, Adams EK, Mackiewicz MR, Weissman TA, Unni VK (2022). Phosphorylation of the aggregate-forming protein alpha-synuclein on serine-129 inhibits its DNA-bending properties. Journal of Biological Chemistry. Dec 29:101552.
Weston LJ, Bowman AM, Osterberg VR, Meshul CK, Woltjer RL, Unni VK (2022). Aggregated Alpha-Synuclein Inclusions within the Nucleus Predict Impending Neuronal Cell Death in a Mouse Model of Parkinsonism. International Journal of Molecular Science. Dec 4;23(23):15294. doi: 10.3390/ijms232315294.
Boutros SW, Raber J, and Unni VK (2021). Effects of Alpha-Synuclein Targeted Antisense Oligonucleotides on Lewy Body-Like Pathology and Behavioral Disturbances Induced by Injections of Pre-Formed Fibrils in the Mouse Motor Cortex. Journal of Parkinson’s Disease. 11(3):1091-1115.
Weston LJ, Cook ZT, Stackhouse TL, Sal MK, Schultz B, Tobias ZJC, Osterberg VR, Brockaway NL, Pizano S, Glover G, Weissman TA, Unni VK (2021). In vivo aggregation of presynaptic alpha-synuclein is not influenced by its phosphorylation at serine-129. Neurobiology of Disease. May;152:105291.
Weston LJ, Stackhouse TL, Spinelli KJ, Boutros SW, Rose EP, Osterberg VR, Luk KC, Raber J, Weissman TA, Unni VK (2021). Genetic deletion of Polo-like kinase 2 reduces alpha-synuclein serine-129 phosphorylation in presynaptic terminals but not Lewy bodies. Journal of Biological Chemistry. Jan 8;100273.
Schaser AJ, Stackhouse TL, Weston LJ, Kerstein PC, Osterberg VR, Lopez CS, Dickson DW, Luk KC, Meshul CK, Woltjer RL, Unni VK. (2020). Trans-synaptic and retrograde axonal spread of Lewy pathology following pre-formed fibril injection in an in vivo A53T alpha-synuclein mouse model of synucleinopathy. Acta Neuropathologica Communications. Aug 28;8(1):150.
Schaser AJ, Osterberg VR, Dent SE, Stackhouse TL, Wakeham C, Boutros SW, Weston LJ, Owen N, Weissman TA, Luna E, Raber J, Luk KC, McCullough AK, Woltjer RL and Unni VK (2019) Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Scientific Reports. 9(1):10919.
Brockway NL, Cook ZT, O'Gallagher MJ, Tobias ZJC, Gedi M, Carey KM, Unni VK, Pan YA, Metz MR, Weissman TA (2019) Multicolor lineage tracing using in vivo time-lapse imaging reveals coordinated death of clonally related cells in the developing vertebrate brain. Developmental Biology. 453(2):130-140.
Volpicelli-Daley LA, Abdelmotilib H, Liu Z, Stoyka L, Daher JP, Milnerwood AJ, Unni VK, Hirst WD, Yue Z, Zhao HT, Fraser K, Kennedy RE, West AB (2016) G2019s-LRRK2 expression augments α-synuclein sequestration into inclusions in neurons. Journal of Neuroscience. 36(28):7415-27.
Osterberg VR, Spinelli KJ, Weston LJ, Luk KC, Woltjer RL, Unni VK (2015) Progressive aggregation of alpha-synuclein and selective degeneration of Lewy inclusion-bearing neurons in a mouse model of parkinsonism. Cell Reports. 10:1252-1260.
Spinelli KJ, Osterberg VR, Meshul CK, Soumyanath A, Unni VK (2015) Curcumin treatment improves motor behavior in alpha-synuclein transgenic mice. PLoS ONE. 10:e0128510.
Spinelli KJ, Taylor JK, Osterberg VR, Churchill MJ, Pollock E, Moore C, Meshul CK, Unni VK (2014) Presynaptic alpha-synuclein aggregation in a mouse model of Parkinson's disease. Journal of Neuroscience. 34:2037-2050.
Ebrahimi-Fakhari D, McLean PJ, Unni VK (2012) Alpha-synuclein's degradation in vivo: Opening a new (cranial) window on the roles of degradation pathways in Parkinson disease. Autophagy. Feb 1;8(2).
Ebrahimi-Fakhari D, Cantuti-Castelvetri I, Fan Z, Rockenstein E, Masliah E, Hyman BT, McLean PJ and Unni VK (2011) Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of α-synuclein. Journal of Neuroscience. 31:14508-14520.
Unni VK, Ebrahimi-Fakhari D, Vanderburg CR, McLean PJ, Hyman BT (2011). Studying
protein degradation pathways in vivo using a cranial window-based approach.
Methods. 2011 53:194-200.
Unni VK, Weissman TA, Rockenstein E, Masliah E, McLean PJ, and Hyman BT (2010)
In Vivo Imaging of α-Synuclein in Mouse Cortex Demonstrates Stable Expression and Differential Subcellular Compartment Mobility. PLoS ONE. 5:e10589.
Lab members

Lab members

Moriah R. Arnold
Program in Biomedical Sciences MD/PhD student
BA, Carleton College, Northfield, MN
I graduated from Carleton College in 2017 with a major in Biology and minors in Neuroscience and Biochemistry. After working at OHSU as a research assistant for 2 years, I matriculated into the OHSU MD/PhD program in 2019. I transitioned from medical school to my PhD in 2021 and joined the Unni Lab where my dissertation project is to investigate the role of alpha-synuclein in melanoma. Although alpha-synuclein is well-recognized to aggregate in neurological disorders like Parkinson’s Disease, it is also highly expressed by melanoma cells for unclear reasons. Interestingly, patients with Parkinson’s Disease and their first-degree relatives are at increased risk of developing melanoma and melanoma patients are at increased risk of Parkinson’s Disease. I hypothesize that melanoma upregulates alpha-synuclein expression to improve DNA repair function and this contributes to protected melanoma growth and metastasis. I hope to match into a research-focused neurology residency program when I graduate from the MD/PhD program in 2027. Outside of science, I enjoy exploring restaurants in Portland, training at my boxing gym, going to Portland Thorns games, and hiking with my partner and dog.

Anna M. Bowman
Neuroscience Graduate Program PhD student
BS, University of Minnesota, Twin Cities, Minneapolis, MN
I graduated with a Bachelor of Science from the University of Minnesota in 2017 with a double major in Neuroscience and Genetics & Cell Biology. From 2018 to 2020, I was a research assistant in the lab of Skylar Jackman at the Vollum Institute at OHSU, where I worked on the contribution of synaptic facilitation to working memory in mice. In the fall of 2020 I entered the Neuroscience Graduate Program at OHSU. I started working in the Unni Lab in the fall of 2021 and collaborate with Dr. Randy Woltjer’s lab in the Pathology Department at OHSU. My interest is in understanding the relationship between alpha-synuclein and beta-amyloid aggregation in the cortex of mice that model Lewy Body Dementia, Parkinson’s Disease Dementia, and Lewy Body Variant Alzheimer’s Disease.

Jessica Keating
Program in Biomedical Sciences MD/PhD student
BA, Mount Allison University, New Brunswick, Canada
MSc, University of Edinburgh, Scotland, UK
I graduated from Mount Allison University in New Brunswick, Canada in 2013 with a BA in Psychology, and from the University of Edinburgh in 2015 with a MSc in Cognitive Neuropsychology. I later worked as an English teacher in South Korea and conducted research with young stroke survivors in Melbourne, Australia, where I made the decision to pursue physician-scientist training. After two subsequent years of pre-med and one year as a research assistant at Rutgers University in New Jersey, I joined OHSU’s MD-PhD program in 2021.
My research focus has evolved from human memory with Jennifer Tomes and Alexa Morcom, to needs assessment and stroke service delivery with Karen Borschmann and Julie Bernhardt, to spinal cord circuits of pain and touch with Victoria Abraira. I now investigate the genetic and molecular modulators of alpha-synuclein’s pathological effects in the central nervous system in the Martin and Unni Labs. When I’m not doing science, you can find me on a PNW mountain or at home making a meal for friends.

Carlos Soto Faguas
Postdoctoral fellow
BS, University of Sevilla, Sevilla, Spain
PhD, Autonomous University of Barcelona, Barcelona, Spain
I finished my undergraduate studies in Basic and Experimental Biomedicine at the University of Seville in 2015, having my first contact in the neuroscience field working with a mouse model of Parkinson’s Disease. After that, I obtained my Ph.D. in neurosciences from the Autonomous University of Barcelona in 2021. My work was focused on the relationship between presenilins, the main genes implicated in familial Alzheimer’s disease, and tau pathology, using transgenic mouse models. In December 2021, I joined Dr. Unni’s lab as a postdoctoral fellow to investigate the role of tau protein in alpha-synuclein aggregate propagation and to study the role of a specific mutation in the ApoE gene in the protection against pathological tau formation in Alzheimer’s Disease. I am looking forward to continuing to do research in neurodegenerative disorders, and specifically, I am highly interested in studying proteinopathies using in vivo imaging techniques. Outside of the lab, I love sports, especially soccer and basketball, traveling, hiking, and hanging out with friends.

Elizabeth P. Rose
Neuroscience Graduate Program PhD student
BA, Pomona College, Claremont, CA
I joined the Neuroscience Graduate Program in the fall of 2019 after graduating from Pomona College double majoring in Neuroscience and Dance. After moving from Southern California and making Portland my new home, I knew I wanted to join a lab studying neurodegenerative diseases. It was a no-brainer to join the Unni Lab in March of 2020. Since then, I have worked on the project studying the role of alpha-synuclein in double-strand break repair. It should be noted that I received the honorable mention of Lab’s Best Western Blot Loading Technique at last year’s Unni Lab Award Ceremony (ULAC). I also serve as the GSO Student Representative on the OHSU SOM Alumni Association council, the 2022–2023 NGP Admissions Committee, and am the founding member of the Unni Lab Party Planning Committee (ULPPC). In my spare time, I attend dance classes at the various studios throughout PDX and am always trying new restaurants and wineries in this Foodie City!

Elias (Eli) Wisdom
Program in Biomedical Sciences MD/PhD student
BS, Pacific University, Forest Grove, OR
I am an MD-Ph.D. student studying the molecular nature of Alzheimer’s Disease, Parkinson’s Disease, and Dementia with Lewy bodies under mentorship from Dr. Vivek Unni, MD, Ph.D., and Andrew Emili, Ph.D. I grew up in La Grande, OR, where I developed a strong appreciation for the natural world and a deep passion for baseball. Following these, I attended Pacific University, in Forest Grove, OR, where I studied Biology while competing on the varsity baseball team all four years. During the summer before my senior year, I completed an internship at Fred Hutchinson Cancer Research Center in Seattle, WA, under mentorship from Harmit Malik, Ph.D., where I first learned that MD-PhD programs existed. Knowing that I could combine careers in medicine and scientific research with this path, I sought out further research and clinical training and moved to New Haven, CT, where I joined the lab of Daniel Colón-Ramos at Yale School of Medicine to study the cell biology of the synapse. My time in the Colón-Ramos Lab and volunteering at Yale New Haven Hospital affirmed my desire to have a career at the intersection of medicine and research. I was fortunate to matriculate into the MSTP program at Oregon Health & Science University in 2021, where I have been able to have outstanding training in his research and clinical interests while living close to family.
Parkinson’s Disease and Alzheimer’s Disease are the two most prevalent neurodegenerative diseases, with little to no treatments available to halt their progression. Having family members and friends afflicted with these diseases, and from my countless clinic visits with patients both before and during medical school, I am motivated to understand the basic biology that drives these conditions, and how to translate this into curative and preventative treatments.
Unni Lab alums
Sydney Weber Boutros, PhD (PhD student 2016-2022)
Current position: Assistant Professor, Boise State University
Zoe Cook, BA (Research assistant 2021)
Current position: Neuroscience Graduate Student, Stanford University
S. Elise Dent, BA (Research assistant 2016-2021)
Current position: Between Positions
Dennisha King, BS (Neuroscience post-baccalaureate 2019-2020)
Current position: Neuroscience Graduate Student, University of Rochester
Meredith Norton, BS (Research assistant 2019-2020)
Current position: Medical Student, University College Dublin
Valerie Osterberg, BA (Research associate 2012-2023)
Current position: Research Associate, Reed College
Allison Schaser, PhD, CCC-SLP (Postdoctoral fellow 2015-2020)
Current position: Assistant Professor, Purdue University
Kateri Spinelli, PhD (Postdoctoral fellow 2012-2015)
Current position: Investigator-initiated Research Manager, Providence Heart Institute
Teresa Stackhouse, BA (Research assistant 2016-2018)
Current position: Neuroscience Graduate Student, Vollum Institute-OHSU
Jonathan Taylor, BS (Research assistant 2012-2014)
Current position: Research Assistant, Pacific University
Leah Weston, MD (Research assistant 2014-2016)
Current position: Internal Medicine Resident, Columbia University