CRISPR/Cas9 System Background
Genome Editing via CRISPR/Cas9 system
Background
The following paragraph is short description from "Mouse Genome Editing Using the CRISPR/Cas System" Donald W. Harms, Rolen M. Quadros, Davide Seruggia, Masato Ohtsuka, Gou Takahashi, Lluis Montoliu, and Channabasavaiah B. Gurumurthy Curr. Protoc. Hum. Genet. 00:15.7.1-15.7.27. 2014
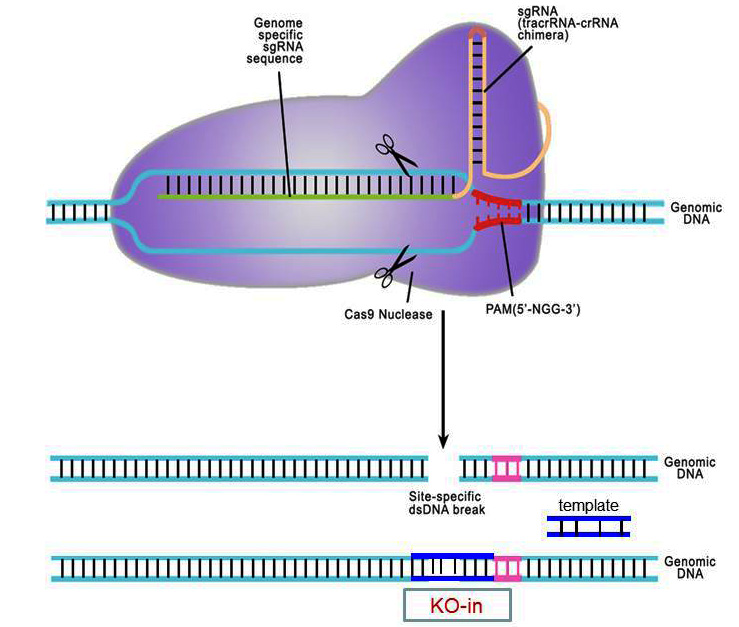
"The CRISPR/Cas system was originally described as an adaptive immune mechanism against invading viruses in bacteria (Barrangou et al., 2007). The system comprises two RNAs and one protein component: a CRISPR RNA (crRNA), a short RNA that undergoes complementary binding with the foreign DNA;a tracrRNA that hybridizes with the crRNA;and a Cas9 enzyme that interacts with the DNA:RNA complexes and cleaves the DNA at a specific site (Horvath and Barrangou, 2010). This system has been redesigned for gene-editing purposes (Cong et al., 2013;Jinek et al., 2013;Mali et al., 2013b;Terns and Terns, 2014) by combining crRNA and tracrRNA into a chimeric unit called a single guide RNA (sgRNA), and by codon-optimizing the Cas9 enzyme sequence to suit mammalian expression. Hence, the CRISPR/Cas9 system used for gene editing consists primarily of two components: a guide RNA that detects the specific sequence in the genome and the Cas9 enzyme that binds to the sgRNA and cleaves the DNA at the target site. While Cas9 mRNA is a common component, the sgRNA is the unique component for each specific gene-editing experiment. When DNA is cleaved, it mainly gets repaired through a mechanism called Non-Homologous End Joining (NHEJ), which is a highly error-prone mechanism that causes a few base pair insertions or deletions (indels) at the cut site. Such an event, in most cases, results in a frame-shift mutation of the coding sequence, eventually leading to gene disruption (a knockout). In cases where a specific mutation is to be introduced at the cut site (called knock-in), a repair template DNA is also needed as a third component of the CRISPR/Cas system, which becomes inserted at the cut site through Homology-Directed Repair (HDR). The repair template DNA can be either a single-stranded oligonucleotide or a double-stranded plasmid/linear DNA. The CRISPR/Cas9 components (sgRNA, Cas9 mRNA with or without a repair DNA) can be introduced into a one-cell-stage mouse embryo to generate offspring that can potentially contain knockout or knock-in mutations. The process involves four major steps: (1) designing CRISPR targets (Basic Protocol 1);(2) synthesis and purification of RNA and DNA components;(3) isolation of one-cell-stage mouse embryos, microinjection of CRISPR/Cas components, and transfer of injected embryos into pseudopregnant mice and (4) genotyping of offspring and sequencing of PCR products to identify mutations."
CRISPR frequently asked questions
CRISPR provides researchers a fast and cost-effective genome-editing tool to modify the genomes of the mouse. The benefits of CRISPR derive from the fact that permits the generation of novel mouse models by gene targeting at the one cell embryo (zygote) stage avoiding the much longer process of modifying and selecting embryonic stem (ES) cells for blastocyst injection and transfer. As described above, this is accomplished with similar flexibility of genome modification as offered by the ES cell approach. Since every embryo is manipulated, there is a higher chance to generate founders.
In general, KO production using CRISPR/Cas9 can create an InDel in 3-4 months, whereas it will take more than one year to produce KO mice by traditional ES cell targeting.
Yes, it is possible to use the system for ES cells and somatic cells in culture (Yang et al., 2014), however as described above most manipulations typically carried out in ES cells can be achieved using CRISPR.
a) Depending on the sequence of the gene, there is a possibility of low flexibility in model design and transgene insertion size.
b) Possible "off target effects" and related unexpected phenotypes, which would need to be identified and bred out in subsequent generations.
At this time, this is not a service offered by the TMM. OHSU users have had success designing and producing their own guide RNAs (gRNA) in combination with commercially produced CAS9 mRNA or protein (see the next question for details). Alternatively, several companies now offer gRNA design and production services. We cannot endorse a particular source, but have listed below a few such options. Keep in mind that some firms offer "off the shelf" gRNAs for disruption of genes, if you are interested in a "knock-in" modification then the site of gRNA cleavage is limited and most likely will require a custom guide.
Yes, but it requires a fair amount of molecular biology experience to generate high quality, highly purified gRNAs in sufficient quantities to permit successful zygotic injections. An alternative to using synthetic guide RNAs is to use a plasmid based system that encodes the Cas9 gene and contains a multiple cloning site for gRNAs that target the gene of interest. This approach requires cloning and does have the caveat that injection of the plasmid delays expression and activity of the Cas9/gRNA complex resulting in an increased possibility of generating chimeric animals.
It is possible to use CRISPR to introduce genomic alterations such as point mutations, epitope tags, fluorescent protein fusions, and other epitope tags. This requires the co-injection of a fragment of "donor" DNA that contains the mutation of interest in the context of the target gene. Flanking sequences guide the replacement of this donor for the wild-type locus. This process is catalyzed by CRISPR directed cleavage and care should taken to use donor DNA that contains significant flanking regions beyond the mutation and that the donor DNA lacks the target site of the gRNA used to generated the genomic cleavage.
CRISPR projects have lower overall costs compared to ES cell based targeting. Moreover, we now have special lower prices for pilot CRISPR projects. See fees for CRISPR/cas9 Microinjections.