Services and Costs
The Proteomics Shared Resource offers several different services to labs, both at OHSU and around the world.
Here are some examples of what we can do for you:
- Protein Identification / Partial Sequencing
- Identification and Localization of Post-Translational Modifications
- Quantitative comparison of protein abundances in complex mixtures using 11-plex TMT labeling
- Identification of protein/protein interactions by analysis of affinity purified complexes
- Targeted MRM analysis on known proteins
- Determination of whole protein mass
- Preparation of submissions for public data repositories
We also advise consulting with us prior to starting an experiment to increase your chances for success. This is especially important for harder experiments, such as identifying Post-Translational Modifications, which can otherwise have a low success rate. An overview of how we perform each of these analyses can be found in the guide to Mass Spectrometry Based Proteomics in the educational portion of our website.
-
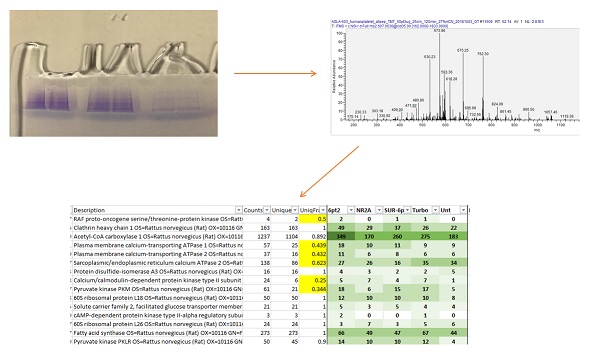
Protein identifications from gel bands Identification of unknown proteins is the bread-and-butter analysis for a Proteomics Facility. Proteins are enzymatically digested into peptides, which are then introduced into a mass spectrometer via a liquid chromatography system. Once inside the mass spectrometer we fragment the peptides to produce MS/MS spectra. These fragmentation patterns are matched against theoretical spectra from a protein database. Scoring algorithms and statistical tools are then used to determine the identity of the proteins in the sample.
Samples can be submitted as either a liquid solution of the protein(s), or as a band from a gel. Liquid samples may also by lyophilized or taken to dryness before submission.
-
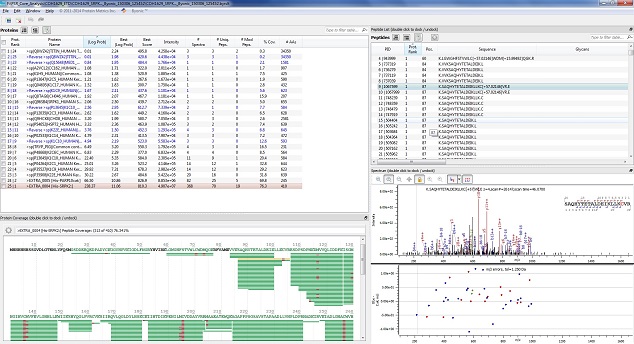
Byonic viewer This type of analysis is a step beyond simple protein identification; identifying Post-Translational Modifications by looking for mass additions in a peptide's MS/MS spectra. The mass shift of the potential PTM is entered into the protein identification software, and the software performs multiple searches of potentially modified peptides looking for forms both with and without the PTM. This analysis can take considerably longer because the search time increases greatly with each potential modification added. In addition the data analysis can be considerably more complicated for this kind of analysis. The end result is a longer and more expensive project, but one that can yield very useful information.
We have several different pieces of software for searching for PTMs:
- Proteome Discoverer provides a basic spectra image, and is used to validate conventional PTMs found via our Comet/PAW pipeline.
- Byonic has a wildcard search for looking for unspecified modifications in a mass range as well as functions for searching for glycosylation. We use it in instances where an exact mass of a modification is unknown, if a peptide is expected to be heavily modified, and for many other unconventional modifications.
- Skyline can be used in conjunction with a targeted analysis to look for cross-linked and disulfide peptide signals.
As with protein identification, samples can be submitted as either a liquid solution of the protein(s), or as a band from a gel. Liquid samples may also by lyophilized or taken to dryness before submission. It's often worth consulting with PSR prior to starting a PTM project, as there are many bench-top procedures with can be done prior to sample submission to increase your odds of a successful project.
-
For simple cases, like in a Co-IP experiment, the relative quantitation of the same protein in different samples is usually performed via spectral counting. This type of analysis is simple and easy to perform, as the number of times MS/MS spectra matches to a protein entry in the database has been shown to be roughly proportional to the amount of protein present. Though the analysis of this data is relatively straight forward it requires that more effort is spent on preparing the samples. Errors introduced in the sample preparation phase can result in serious problems with the analysis.
Samples for this kind of analysis are usually submitted as either a liquid solution, or lyophilized powder, which we subsequently run into an SDS-page gel prior to digestion and analysis. For a more complete description of the ins and outs of this experiment be sure to check out our Co-IP guide.
-
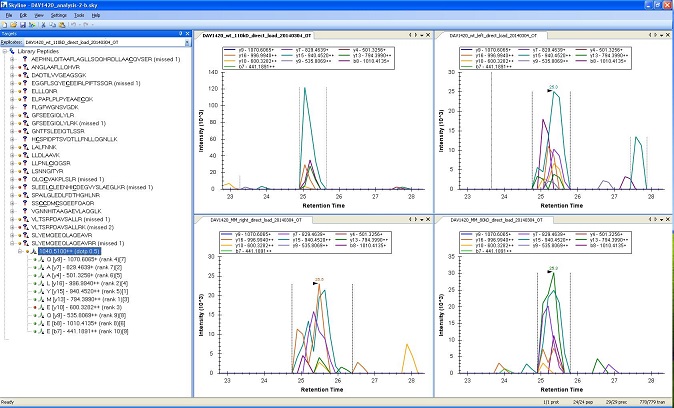
Targeted MRM analysis Multiple Reaction Monitoring analysis is a way to do good quantitation on proteins with a relatively low amount of background interference. However a lot of groundwork must be completed before a MRM Analysis can take place.
These experiments usually take significant amounts of time and resources for the initial development of the assay. The masses chosen for monitoring must be determined before the experiment, and this can require several mass spectrometry runs to gather a list of ions to attempt the experiment on. Subsequent runs assess the different potential ion transitions, and weed out less robust candidates. This is in addition to any benchwork concerns, as creating consistent samples is extremely important. However once all this is complete the costs to repeat an assay is minimal, and is this low repeat cost is the common motivating factor for these experiments.
If you'd like to attempt an MRM experiment, close consultation with the Proteomics Shared Resource is absolutely necessary. If you'd like to perform this type of analysis please contact PSR, so that we can assist you in this process.
-
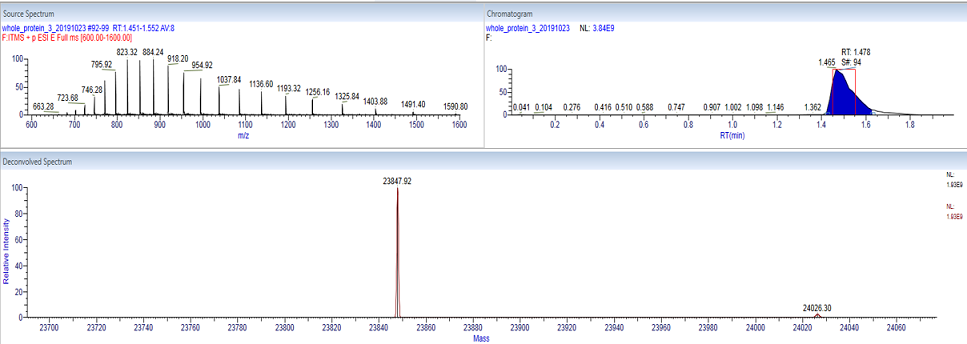
Protein Whole Mass Deconvolution Whole mass determination of a protein is often a relatively straight-forward analysis. When the protein is introduced into the mass spectrometer a series of charge states are produced. Deconvolution software can interpret this complex spectrum, and reconstruct the mass of the protein.
The image to the left shows the measurement of a recombinantly expressed protein with a mass of 23,848. The multiple charge states of the protein (upper left) were deconvoluted to create the mass spectrum (bottom). The region in the red box shows the time interval where acquired spectra were averaged for the deconvolution. The approximate expected mass of the protein being analyzed should be given ahead of time during submission to aid in the data workup. The deconvolution of the results below was performed using Thermo Scientific Deconvolution 4.0 software.
-
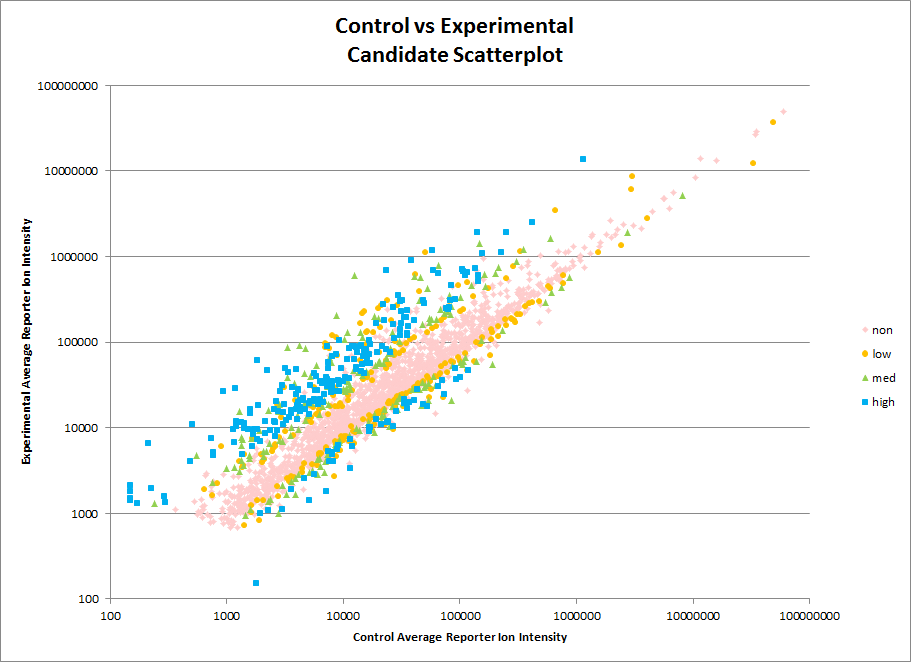
Differential candidates on a scatterplot Our Orbitrap Fusion is set up to do quantitative analysis using TMT tags. This kind of analysis can be used to track changes in protein abundance across multiple samples for thousands of proteins. As always consultation with PSR is encouraged before starting an experiment. Considerations for TMT samples include numbers of replicates and statistical methods to be used for analysis. Costs can vary considerably depending on the scope of the work. For some examples of the costs of a TMT analysis see our Rates page.
-
The submission of data files to ProteomeXchange is a requirement for nearly all publications involving Proteomics data. There are a number of different pieces of information that are required for the submission, and the template form below lists the different fields that will need to be filled out. In general it’s best to prepare all the information for submission before using the PRIDE uploader, as it is difficult to edit text in the small windows provided. Also special characters, such as Greek letters, will have to be removed prior to submission.
Many of the required details can be taken directly from the manuscript, but there may be additional information needed that isn’t in the publication. It will also be necessary to upload final results files, raw data files, the .fasta database used, and other intermediate files created during the analysis. PSR keeps all these files archived here for several years after your analysis, and is happy to help prepare the submission, and upload the data as well. Depending on the size and scope of what's needed there may be charges involved, as gathering and preparing the data for upload can take several hours.